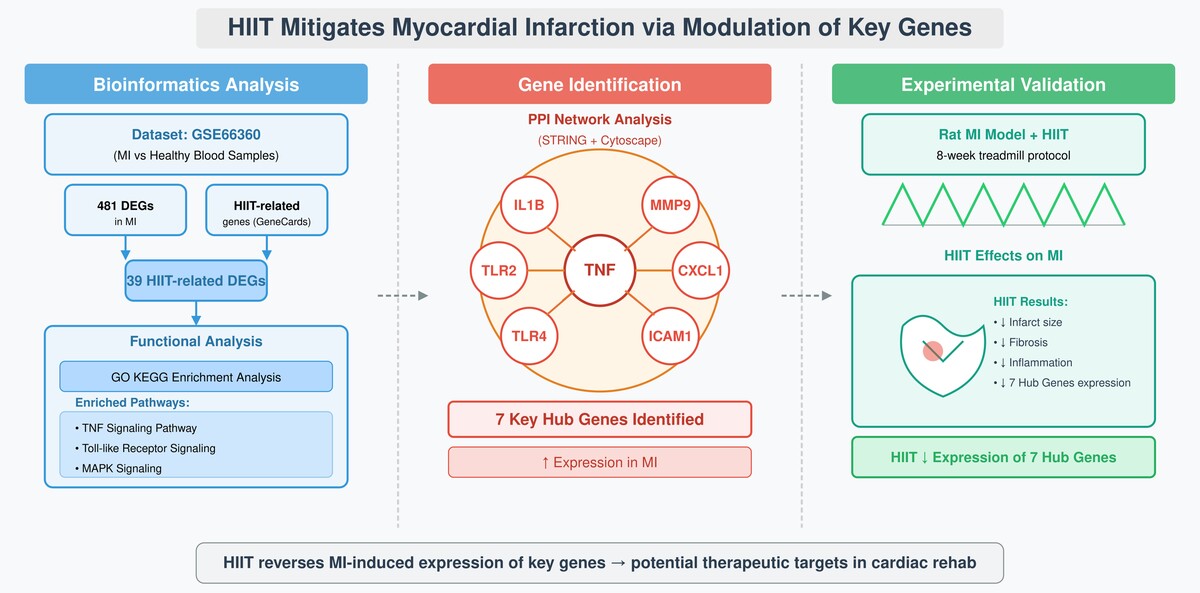
Introduction
Cardiovascular disease (CVD) remains the primary cause of death worldwide and is responsible for 17.9 million deaths annually [1]. Myocardial infarction (MI), a common and severe complication of coronary artery disease (CAD), is caused by coronary artery occlusion, with sudden loss of oxygen supply due to decreased or complete cessation of blood flow to the myocardium, leading to heart failure and millions of deaths each year [2, 3]. While myocardial reperfusion, such as percutaneous coronary intervention, is the mainstay of MI treatment, it can also cause irreversible cardiomyocyte injury and further necrosis [4, 5]. Therefore, there is a critical need to explore adjunctive therapies that can mitigate MI progression and improve patient outcomes by targeting the underlying molecular mechanisms.
Cardiac rehabilitation (CR) is a cornerstone of CAD management, significantly improving the prognosis of MI patients through exercise-based interventions [6]. High-intensity interval training (HIIT), defined as intermittent bouts of high-intensity exercise interspersed with low-intensity recovery periods, has emerged as a particularly effective strategy for enhancing cardiorespiratory fitness, as measured by peak oxygen consumption (VO2 peak) [7, 8]. Clinical trials have shown that HIIT outperforms moderate-intensity continuous training in improving VO2 peak and cardiac function in CAD patients [9–11], while animal studies have demonstrated that HIIT reduces infarct size, improves left ventricular function, and decreases cardiac fibrosis in MI rat models [12, 13]. Moreover, HIIT has been shown to ameliorate metabolic syndrome, body composition, and cardiac remodeling in MI patients during early outpatient CR, significantly enhancing quality of life and cardiopulmonary health [14–16]. Despite these benefits, the molecular mechanisms underlying HIIT’s cardioprotective effects in MI remain poorly understood, limiting its optimization as a therapeutic strategy.
To address this gap, we aimed to identify key genes and pathways modulated by HIIT in the context of MI, using an integrated approach combining bioinformatics analysis and experimental validation. By analyzing gene expression profiles from MI patients and healthy controls, intersecting these with HIIT-related genes, and validating our findings in both human samples and MI rat models, we sought to uncover novel molecular targets that mediate HIIT’s protective effects.
This study aimed to deepen our understanding of HIIT’s molecular mechanisms in MI therapy, providing a foundation for developing targeted interventions and guiding future research to optimize cardiovascular outcomes.
Material and methods
Overview of study design
To investigate the molecular mechanisms of HIIT in MI, we employed a multi-step approach: (1) bioinformatics analysis to identify HIIT-related differentially expressed genes (DEGs) in MI, (2) functional enrichment and protein-protein interaction (PPI) analyses to pinpoint hub genes, and (3) experimental validation in human samples and MI rat models to confirm the relevance of these genes. This integrated strategy was chosen to bridge the gap between computational predictions and biological validation, ensuring a comprehensive understanding of HIIT’s effects.
Data acquisition and processing
We selected the GSE66360 microarray dataset from the Gene Expression Omnibus (GEO) database for comprehensive gene expression profiling of MI patients and healthy controls. This dataset, retrieved using the GEOquery R package, includes 49 samples from patients with MI and 50 samples from healthy individuals, analyzed on the GPL570 platform ([HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array). The GSE66360 dataset specifically profiles mRNA expression in circulating endothelial cells, which are relevant to systemic inflammation and vascular responses in MI, aligning with our focus on inflammatory hub genes. To identify genes associated with HIIT, we queried the GeneCards database (https://www.genecards.org/) using the exact search term “high-intensity interval exercise” (with quotations) to ensure precise matching, which yielded 717 relevant genes. This specific keyword was chosen to capture genes linked to physiological adaptations and responses induced by HIIT, ensuring relevance to the study’s focus on exercise-based interventions.
Screening HIIT-related DEGs
Differentially expressed genes (DEGs) between the MI and control groups in the GSE66360 dataset were identified using the limma R package. We applied stringent criteria of |log2 fold change| > 1 and an FDR-adjusted p-value < 0.05 to select genes with biologically meaningful expression changes while controlling for multiple testing. The FDR correction was used to mitigate the risk of type I errors due to multiple comparisons in the bioinformatics analysis, though we acknowledge that the small validation sample sizes may limit detection of smaller effect sizes. The resulting DEGs were visualized through a heatmap and volcano plot generated using the pheatmap, ggplot2, and ggrepel R packages to illustrate the expression patterns and statistical significance. This step allowed us to identify MI-specific molecular alterations that could be modulated by HIIT.
Gene set enrichment analysis (GSEA)
To explore the biological significance of the identified DEGs, we conducted a gene set enrichment analysis (GSEA) using the clusterProfiler R package. The C2 KEGG gene sets from the Molecular Signatures Database (MSigDB), accessed via the msigdbr R package, served as a reference. Pathways were deemed significantly enriched if they exhibited a Normalized Enrichment Score (NES)| > 1 and p < 0.05, providing insight into the molecular pathways altered in MI. GSEA was chosen to contextualize the DEGs within broader biological processes, guiding our subsequent focus on inflammatory pathways.
Functional enrichment analyses
Functional enrichment of HIIT-related DEGs was performed using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses, implemented through the clusterProfiler R package with human genome annotations from the org.Hs.eg.db R package. Significance thresholds were set at p < 0.05 and q < 0.05 to ensure robust control of the false discovery rate (FDR). The results were visualized using the enrichplot and ggplot2 R packages to elucidate the functional roles and pathway involvement of these genes in the context of HIIT and MI. This analysis helped identify potential mechanisms through which HIIT exerts its cardioprotective effects.
PPI construction and selection of hub genes
To investigate interactions among HIIT-related DEGs, we constructed a PPI network using the STRING database (www.string-db.org) with a minimum interaction score of 0.400, which was selected to balance specificity and sensitivity. The network was analyzed in Cytoscape software, where the NetworkAnalyzer tool calculated node degrees to assess connectivity, and the CytoHubba plugin employed the Maximal Clique Centrality (MCC) method to identify hub genes, highlighting key players in the network.
Patient sample collection
Peripheral blood samples were obtained from 20 patients with MI within 12 h of symptom onset and 20 age- and sex-matched healthy donors upon admission. To reiterate, sample collection occurred within 12 h of MI symptom onset to capture acute changes in gene expression. Patients were included if they exhibited confirmed ST-segment elevation on ECG and were excluded if they had a history of inflammatory disorders, cancer, active infections, or conditions that could confound their gene expression profiles. The healthy controls had no history of cardiovascular or chronic disease. A sample size of 20 per group was determined via power analysis, ensuring 80% power to detect a 1.5-fold change in gene expression with a standard deviation of 0.5 at a significance level of 0.05 using G*Power software. All participants provided informed consent, and the study was approved by the Institutional Review Board of Guangxi Normal University, adhering to the principles of the Declaration of Helsinki. The clinical characteristics of participants are shown in Table I.
Table I
Clinical information of healthy controls and MI patients
[i] BMI – body mass index, BUN – blood urea nitrogen, CK-MB – creatine kinase-myocardial band, CRP – C-reactive protein, LVEF – left ventricular ejection fraction, RVEF – right ventricular function. Comparisons were made using the χ2 test for categorical variables and Student’s t test for continuous variables.
RT-qPCR
Total RNA was extracted from the blood samples using TRIzol reagent (Invitrogen, USA) and reverse transcribed into cDNA using a DNA reverse transcription assay kit (Thermo Fisher, MA, USA). Quantitative PCR was performed using TransStart Top Green qPCR SuperMix (Transgen, Beijing, China) on a Bio-Rad CFX96 system (CA, USA). Gene expression levels were normalized to those of GAPDH and quantified using the 2–ΔΔCt method for qPCR. Primer sequences for the hub genes are detailed in Supplementary Table SI, and PCR cycling conditions consisted of an initial denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min, ensuring reproducible and accurate amplification. This method was chosen to validate the bioinformatics findings in a clinical context, focusing on the hub genes identified as potential mediators of HIIT’s effects.
Animal experiment
To test the functional role of the identified hub genes, we conducted experiments in an MI rat model. Male SD rats (7–8 weeks, 270–300 g) were sourced from Vital River (Beijing, China) and maintained under controlled conditions (24 ±2°C, 55 ±5% humidity, 12/12-hour light/dark cycle). Following a 7-day acclimatization period, the rats were randomly assigned to three groups: sham, MI, and MI + HIIT (n = 8 per group). The sample size was determined to provide 80% power to detect a 20% difference in infarct size with a standard deviation of 10% at a significance level of 0.05 based on prior literature [17]. MI was induced by ligating the left anterior descending artery (LAD) 2 mm distal from its origin with a 6.0 silk suture under sodium thiopental anesthesia (50 mg/kg), which induced deep sedation and unconsciousness. Sham rats underwent the same procedure without ligation. The sham and MI groups were sacrificed 2 weeks after surgery to assess acute MI effects, while the MI + HIIT group underwent a 2-week recovery period followed by 8 weeks of HIIT before sacrifice at the end of the 10-week study period. Following the induction of anesthesia, the animals were sacrificed by cardiac perfusion, a method chosen to minimize suffering and preserve tissue integrity for subsequent analysis. The HIIT protocol began with 5 min/day at 10 m/min (40–50% VO2max) for 5 days in week 1, followed by 8 weeks of two daily sessions of 7 min at 25 m/min (85–90% VO2max) and 3 min at 15 m/min (50–60% VO2max), 5 days/week. This protocol was adapted from established HIIT models in MI rats, which typically use high-intensity bouts at 85–90% VO2max interspersed with recovery periods to mimic clinical HIIT interventions [13, 14]. The intensity and duration were chosen to balance efficacy and tolerability in post-MI rats, as higher intensities have been shown to improve cardiac function without exacerbating injury [13]. The rats were sacrificed at the end of the study, and their hearts were excised for further analysis by researchers blinded to group allocation. All procedures were approved by the Laboratory Animal Ethics Committee of Guangxi Normal University and complied with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Triphenyltetrazolium chloride (TTC) staining
The myocardial infarct size was evaluated using TTC staining. Rat hearts were frozen in liquid nitrogen for 20 s, sectioned into 1–2 mm slices, and incubated in PBS containing 2% TTC solution (Sigma-Aldrich, USA) at 37°C for 30 min. Sections were fixed in 4% paraformaldehyde for 24 h and images were captured to distinguish infarcted (white) from non-infarcted (red) tissue.
Histological examination
Heart samples were fixed in 4% paraformaldehyde for 24 h, embedded in paraffin, and sectioned into 5-µm slices. HE and Masson staining were performed using standard kits (Solarbio, Beijing, China) to assess tissue morphology and fibrosis following the manufacturer’s protocols.
Western blot
Proteins were extracted from cardiac tissues using RIPA lysis buffer (Sangon Biotech, Shanghai, China) and quantified using a bicinchoninic acid (BCA) assay kit (Pierce, USA). Proteins were separated on 10% SDS-PAGE gels and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were blocked with 5% skim milk and incubated overnight at 4°C with primary antibodies against TNF (ab307164, 1 : 1000, Abcam), IL1B (ab283818, 1 : 1000, Abcam), MMP9 (ab283575, 1 : 1000, Abcam), TLR4 (SAB5700798, 1 : 1000, Sigma-Aldrich), ICAM1 (SAB5700809, 1 : 1000, Sigma-Aldrich), TLR2 (SAB5701209, 1 : 1000, Sigma-Aldrich), CXCL1 (PA5-115328, 1 : 1000, Thermo Fisher), and GAPDH (ab181602, 1 : 10000, Abcam) as a loading control, followed by HRP-conjugated secondary antibodies for 1 h at room temperature. Protein bands were visualized using enhanced chemiluminescence and quantified using ImageJ software.
Statistical analysis
Data were analyzed using R (version 4.4.1) and GraphPad Prism 8. The Shapiro-Wilk test was used to check the normal distribution of data. For normally distributed data, comparisons between two groups were assessed with Student’s t-test, while multiple group comparisons were conducted using one-way ANOVA with Tukey’s post-hoc test to determine specific differences. Continuous variables are expressed as mean ± SD. Categorical variables are expressed in counts and proportions (%) and compared between two groups using the χ2 test. For non-normally distributed data, the Wilcoxon test was used to compare quantitative variables (gene expression) between groups, with results shown as medians and interquartile ranges (IQRs). Statistical significance was set at p < 0.05.
Results
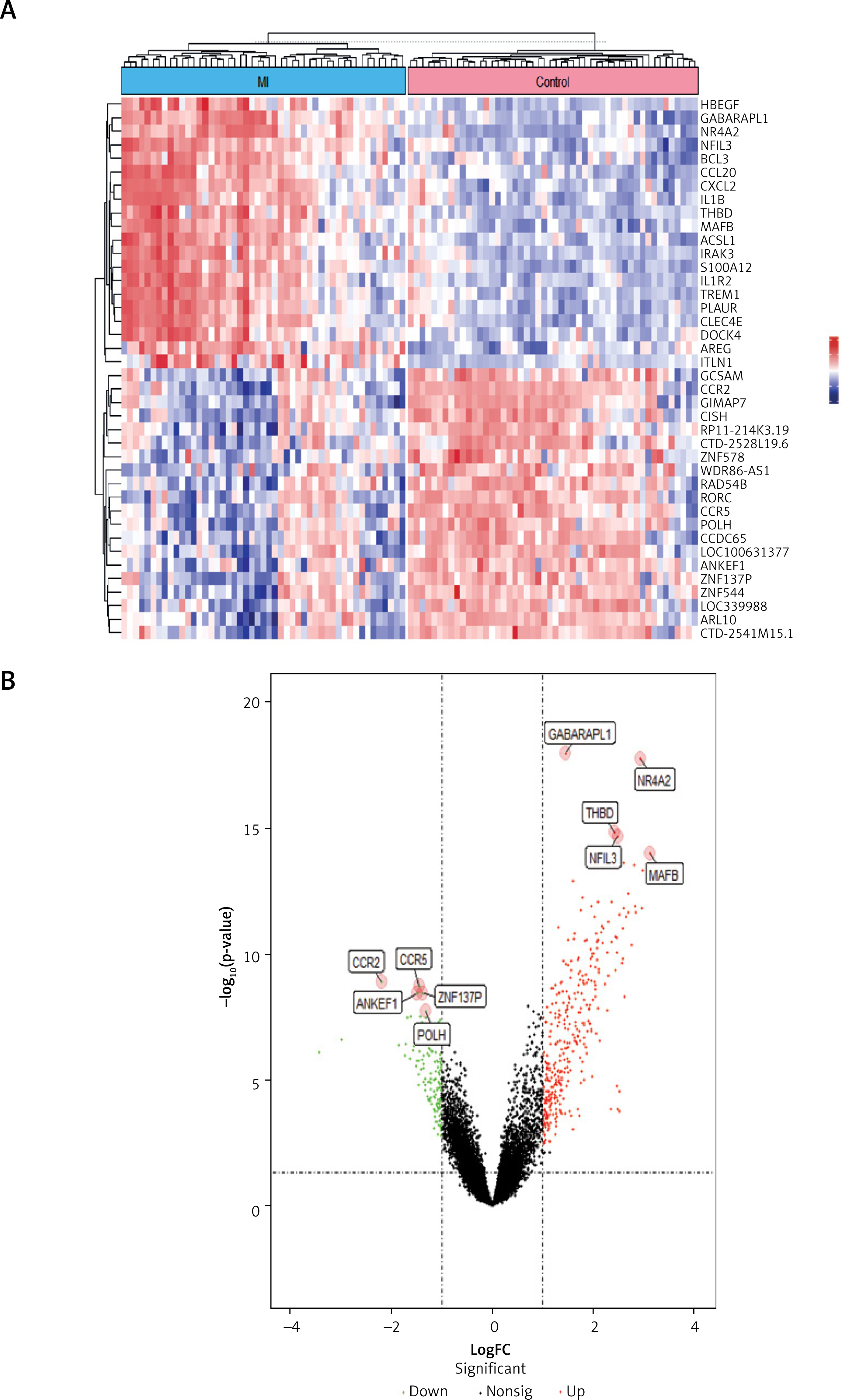
Identification of DEGs in myocardial infarction We first investigated gene expression patterns in circulating endothelial cells from MI patients (n = 49) and healthy controls (n = 50) using the GSE66360 dataset. Differential expression analysis with the limma R package identified 481 DEGs, comprising 351 upregulated and 130 downregulated genes in MI patients compared to controls (FDR-adjusted p < 0.05, |log2 fold change| > 1; Figures 1 A, B). A heatmap of the top 20 significantly upregulated and downregulated genes (Figure 1 A) revealed distinct expression profiles between MI and control groups, with upregulated genes such as GABARAPL1, CCL20, and MAFB, and downregulated genes including LOC10031377, ZNF544, and ARL10. The volcano plot (Figure 1 B) further confirmed the statistical significance of these DEGs, highlighting key upregulated genes such as NR4A2 (logFC = 3.8, –log10 p-value = 15) and MAFB (logFC = 2.5, –log10 p-value = 18), and downregulated genes such as ZNF317P (logFC = –2.1, –log10 p-value = 10) and POLH (logFC = –1.9, –log10 p-value = 8). These DEGs reflect molecular alterations in MI, particularly in inflammatory and immune response pathways, as evidenced by the upregulation of CCL20 and MAFB, which are known to mediate chemokine signaling and macrophage activation in cardiovascular disease, respectively. These findings guided our subsequent pathway analyses to explore the biological significance of these alterations.
Figure 1
Identification of differentially expressed genes (DEGs) in myocardial infarction (MI). A – Heatmap illustrating the top 20 significantly upregulated and downregulated genes in MI patients compared to healthy controls from the GSE66360 dataset. B – Volcano plot displaying the distribution and statistical significance of DEGs between MI patients and healthy controls
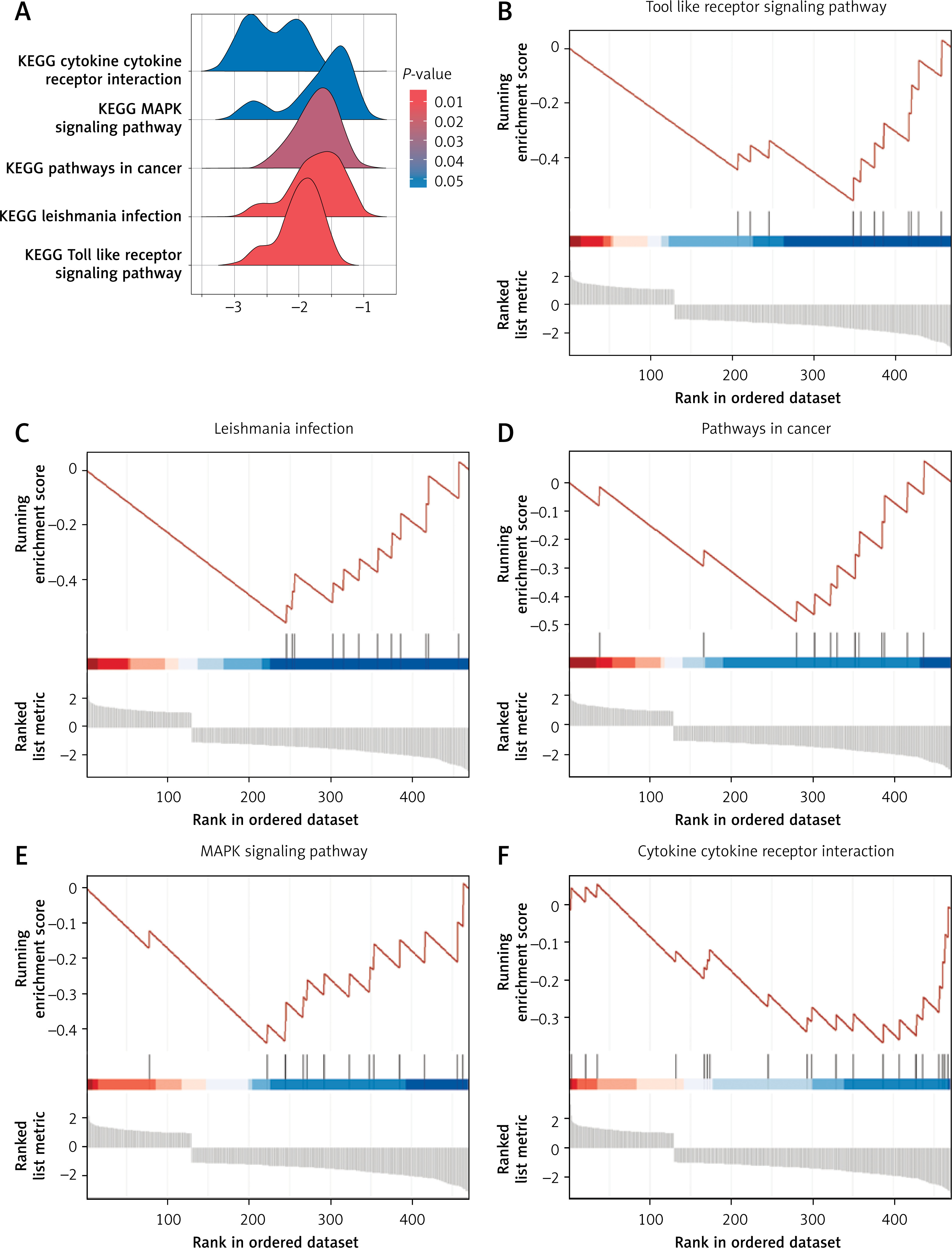
Suppressed inflammatory signaling pathways revealed by GSEA
Gene set enrichment analysis (GSEA) of the 481 DEGs revealed five significantly enriched KEGG pathways (Figure 2 A), all exhibiting negative enrichment scores, indicating downregulation in MI patients relative to healthy controls. These included the Toll-like receptor signaling pathway, Leishmania infection, pathways in cancer, MAPK signaling pathway, and cytokine-cytokine receptor interaction (Figures 2 B–F). The negative enrichment suggests potential suppression or dysregulation of these immune and signaling pathways in circulating endothelial cells during MI, possibly reflecting an impaired systemic immune response or altered cellular signaling. These pathway alterations guided the downstream focus on inflammation-related genes and their modulation by HIIT.
Figure 2
Gene set enrichment analysis (GSEA) of DEGs in MI patients. A – Overview of the five most significantly enriched KEGG pathways identified by GSEA comparing MI patients and healthy controls. B–F – Detailed enrichment plots for selected pathways: B – Toll-like receptor signaling pathway, C – Leishmania infection pathway, D – Pathways in cancer, E – MAPK signaling pathway, F – Cytokine-cytokine receptor interaction pathway
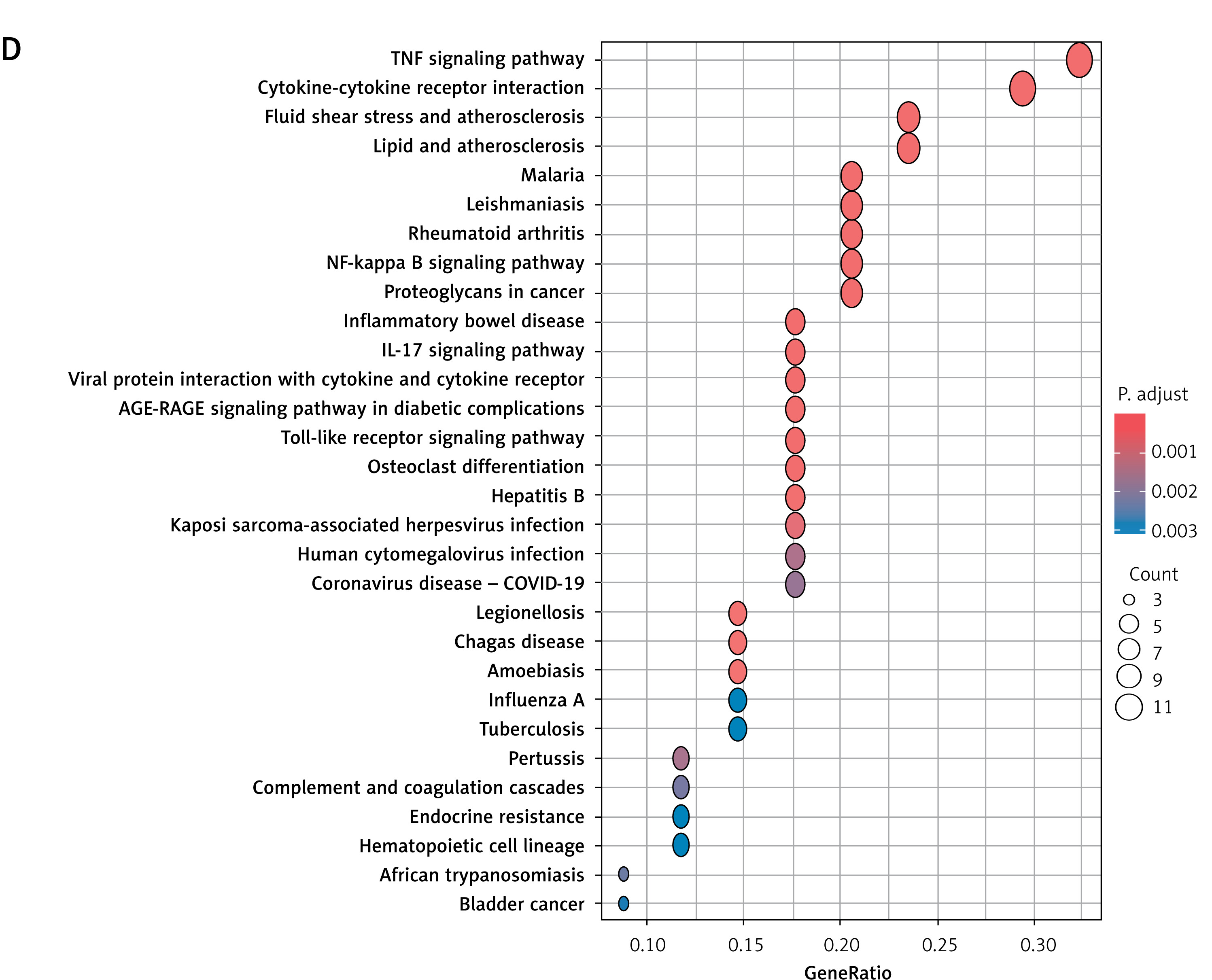
Pinpointing HIIT-responsive genes linked to MI
Intersection of the 717 HIIT-associated genes from GeneCards with the 481 MI-related DEGs identified 39 overlapping genes (Figure 3 A). Correlation analysis revealed predominantly positive co-expression patterns among these genes in MI samples (Figure 3 B). Gene Ontology (GO) enrichment analysis highlighted their involvement in biological processes such as positive regulation of nitric oxide biosynthetic process, nitric oxide metabolic process, reactive nitrogen species metabolic process, and response to lipopolysaccharide (Figure 3 C). Cellular component terms included external side of plasma membrane, secretory granule lumen, and platelet alpha granule lumen, while molecular functions encompassed cytokine receptor binding and *interleukin-1 receptor binding* (Figure 3 C). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis identified significant enrichment in TNF signaling pathway, cytokine-cytokine receptor interaction, Toll-like receptor signaling pathway, NF-κB signaling pathway, AGE-RAGE signaling pathway in diabetic complications, and *IL-17 signaling pathway* (Figure 3 D). These findings underscore the role of HIIT-modulated genes in inflammatory and vascular regulatory mechanisms, particularly through nitric oxide metabolism and innate immune signaling.
Figure 3
Identification and functional analysis of HIIT-related DEGs in MI. A – Venn diagram illustrating the intersection of 39 genes identified as HIIT-related targets (from GeneCards database) and DEGs from the GSE66360 dataset. B – Heatmap demonstrating correlation patterns among the expression levels of these 39 HIIT-related DEGs in MI samples. C, D – Functional enrichment analyses of the selected 39 genes: C – Gene Ontology (GO) analysis highlighting key biological processes, cellular components, and molecular functions, C, D – Functional enrichment analyses of the selected 39 genes: D – KEGG pathway analysis identifying significantly enriched signaling pathways
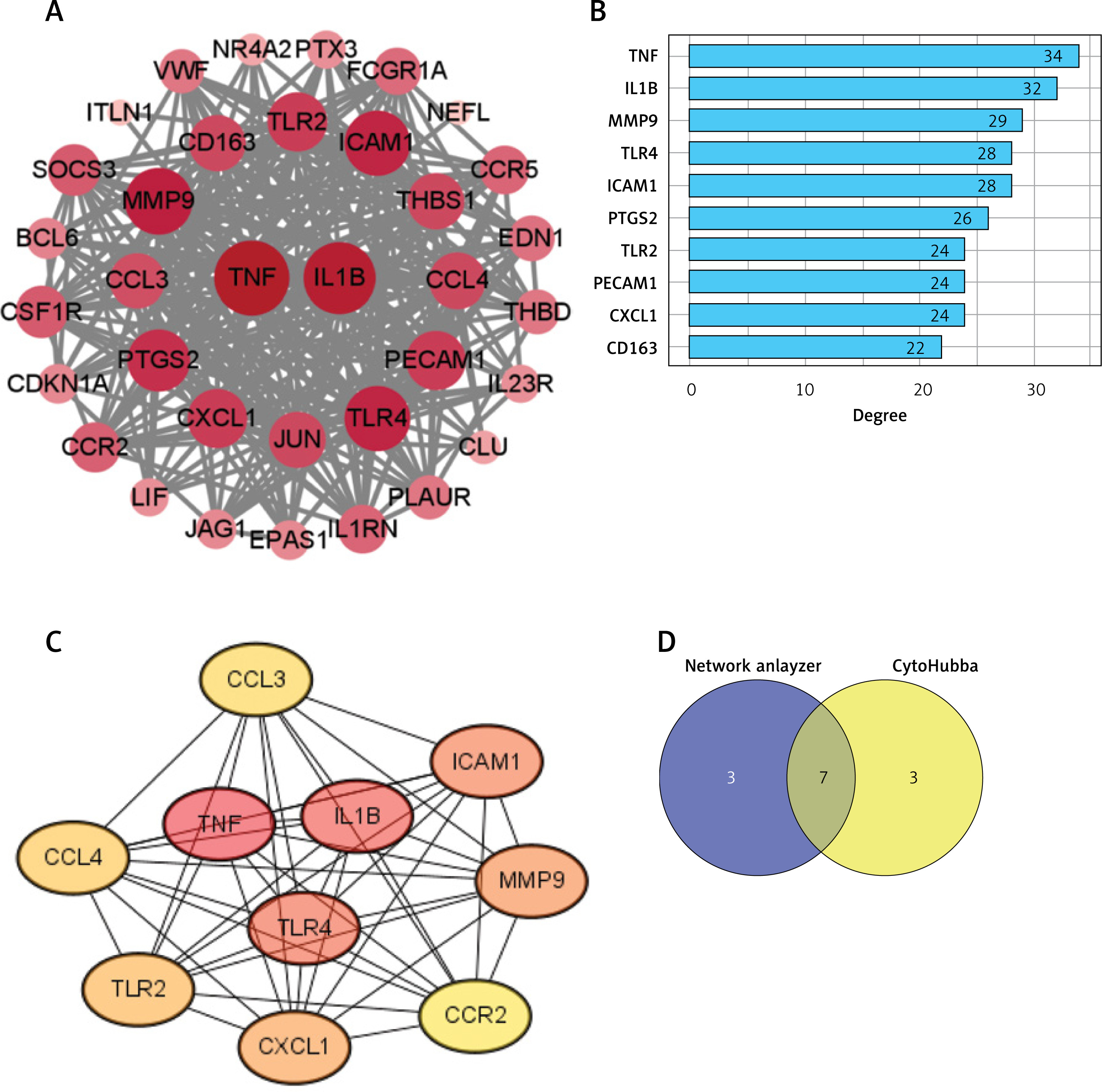
Core inflammatory regulators identified through protein interaction mapping
A PPI network was constructed for the 39 HIIT-related DEGs using STRING and analyzed in Cytoscape, which consisted of 35 nodes and 306 edges after the removal of disconnected nodes (Figure 4 A). NetworkAnalyzer identified TNF and IL1B as the most connected nodes in the network (Figure 4 B). The CytoHubba MCC method ranked TNF, IL1B, TLR4, ICAM1, MMP9, CXCL1, and TLR2 as the top hub genes (Figure 4 C). The intersection of these analyses led to the confirmation of seven hub genes – TNF, IL1B, MMP9, TLR4, ICAM1, TLR2, and CXCL1 (Figure 4 D). These hub genes, which play critical roles in inflammation and immune regulation, align with their known functions in myocardial infarction (MI) pathology. They were subsequently selected for experimental validation to verify their modulation by HIIT.
Figure 4
PPI network construction of the HIIT-related DEGs. A – Visualization of the constructed PPI network for HIIT-related DEGs using STRING database and Cytoscape software. B, C – Ranking analyses identifying critical hub genes: B – Top 10 genes ranked by node degree using NetworkAnalyzer; C – Top 10 genes ranked by Maximal Clique Centrality (MCC) method using CytoHubba plugin. D – Venn diagram showing overlap between NetworkAnalyzer and CytoHubba analyses, identifying seven key hub genes
Seven hub genes confirmed as MI-linked inflammatory drivers
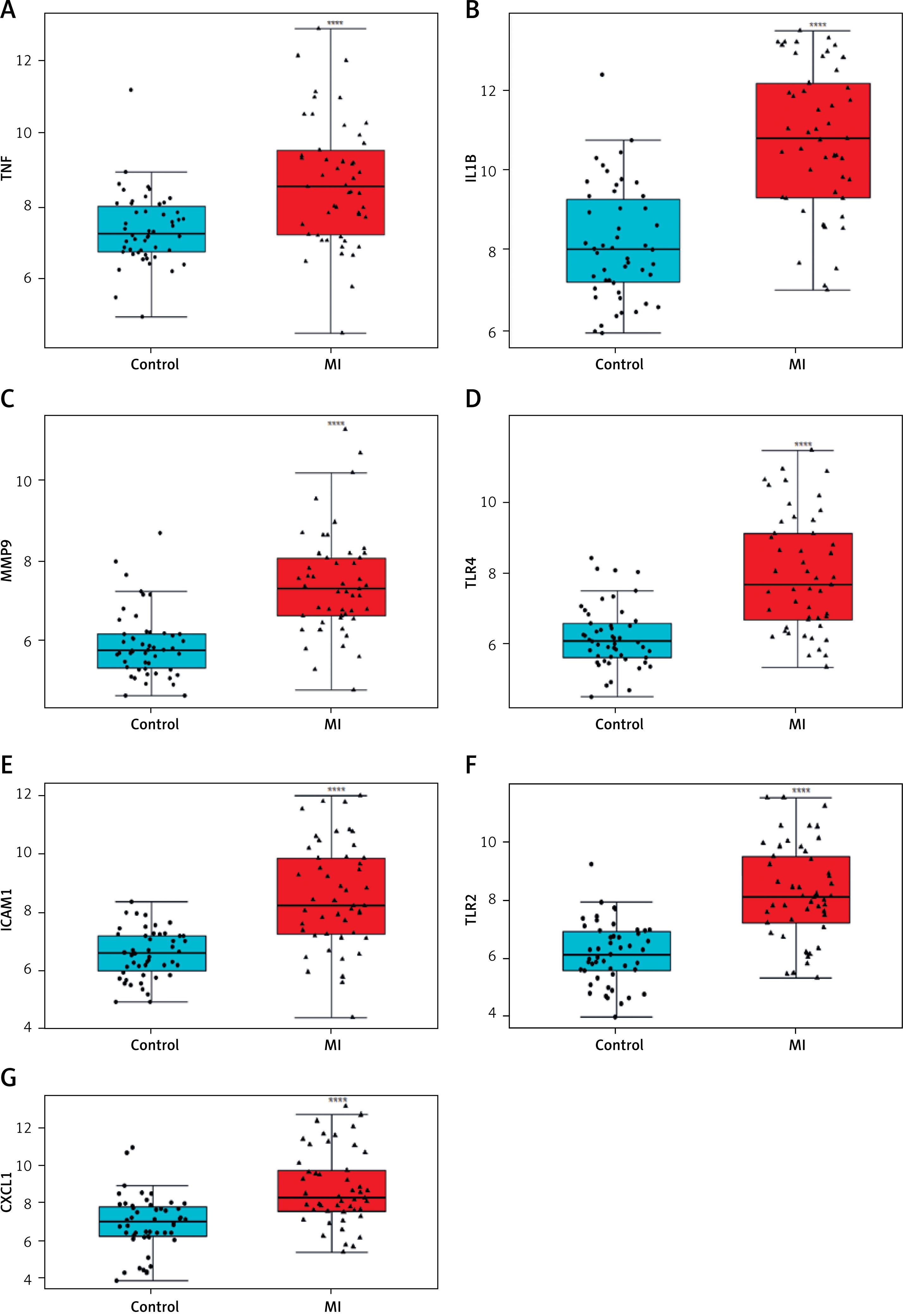
Differential expression analysis using the GSE66360 dataset revealed significant upregulation of all seven hub genes – TNF, IL1B, MMP9, TLR4, ICAM1, TLR2, and CXCL1 – in myocardial infarction (MI) samples compared to controls (p < 0.0001 for all; Figures 5 A–G). Boxplot distributions demonstrated elevated median expression levels and broader variability in MI samples, consistent with inflammatory activation. These results were subsequently validated in an independent cohort via RT-qPCR (Figures 6 A–G), corroborating the increased transcriptional levels of these genes in peripheral blood from MI patients relative to healthy donors (p < 0.001). Together, these findings underscore the clinical relevance of these hub genes as robust biomarkers of MI-associated inflammation.
RT-qPCR confirms robust overexpression of inflammatory markers in MI patients
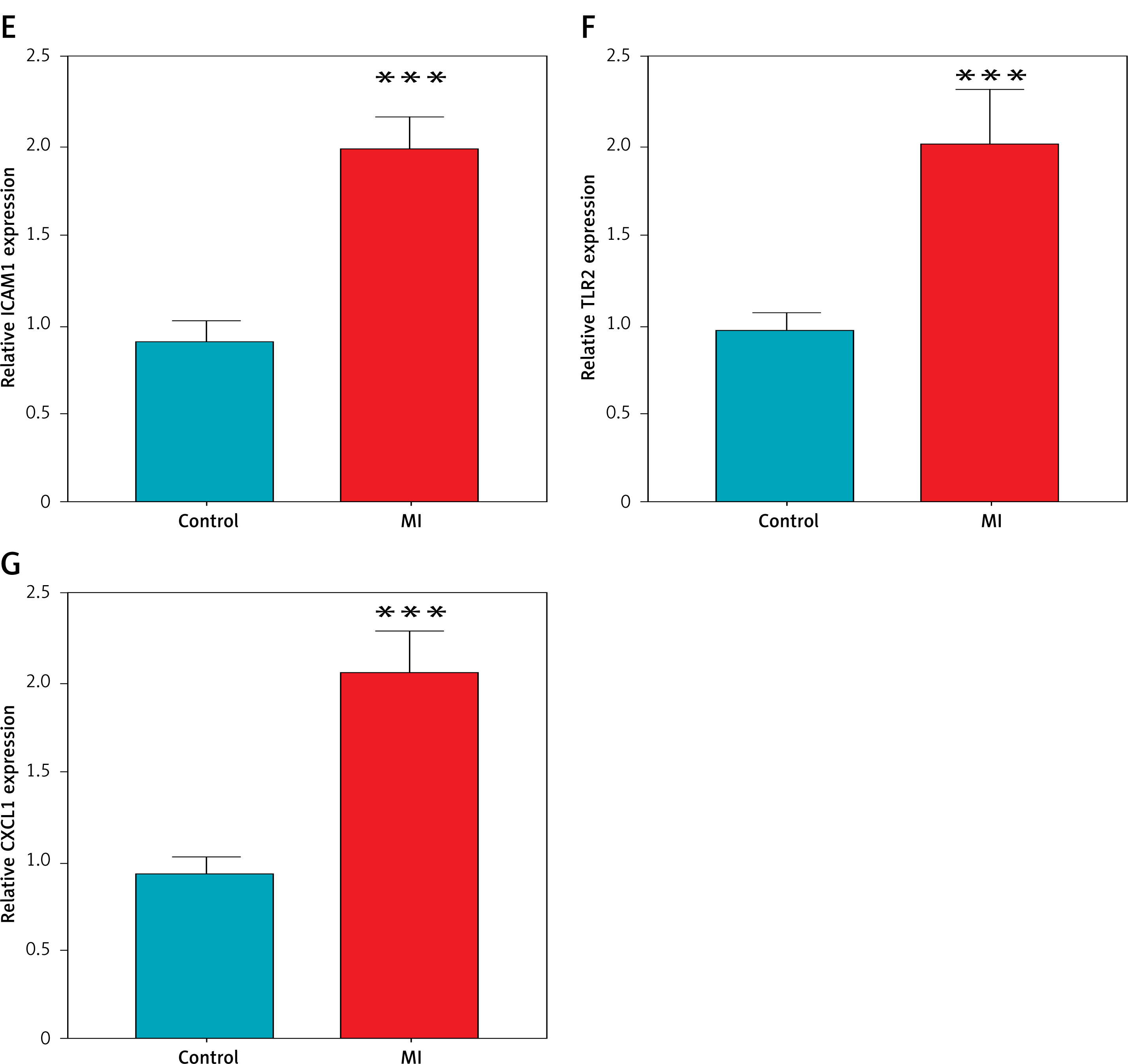
To validate the bioinformatic findings, RT-qPCR was performed on peripheral blood samples from patients with myocardial infarction (MI) and age-matched healthy controls. As shown in Figures 6 A–G, the relative mRNA expression levels of all seven hub genes – TNF, IL1B, MMP9, TLR4, ICAM1, TLR2, and CXCL1 – were significantly elevated in the MI group (***p < 0.001 for all comparisons). These results corroborate the dataset-derived expression trends and further confirm the association of these inflammatory genes with acute MI in a clinical setting.
Figure 6
Experimental validation of hub gene expression in clinical samples by RT-qPCR. Quantitative RT-PCR validation demonstrating significantly elevated mRNA expression levels of hub genes: A – TNF, B – IL1B, C – MMP9, D – TLR4; E – ICAM1, F – TLR2, G – CXCL1 in peripheral blood samples from MI patients compared to healthy donors. ***P < 0.001
HIIT diminishes infarct size and inflammatory gene expression in MI rat models
To assess the impact of HIIT on myocardial infarction (MI) pathology, we evaluated infarct size, myocardial histopathology, and protein expression of inflammatory hub genes in rat models. Triphenyltetrazolium chloride (TTC) staining revealed extensive infarcted areas (pale regions) in the MI group, which were markedly reduced in the MI + HIIT group, indicating a significant reduction in infarct size (p < 0.01; Figure 7 A).
Figure 7
HIIT reduces myocardial injury and hub gene expression in an MI rat model. A – Representative images of myocardial infarct size assessed by triphenyltetrazolium chloride (TTC) staining in sham-operated rats, untreated MI rats, and MI rats subjected to HIIT intervention. B – Histological assessment using Masson’s trichrome staining for fibrosis detection and hematoxylin-eosin staining for morphological evaluation across experimental groups. C – Western blot analysis quantifying protein expression levels of the seven hub genes-TNF, IL1B, MMP9, TLR4, ICAM, TLR2, CXCL1 in cardiac tissues from each experimental group
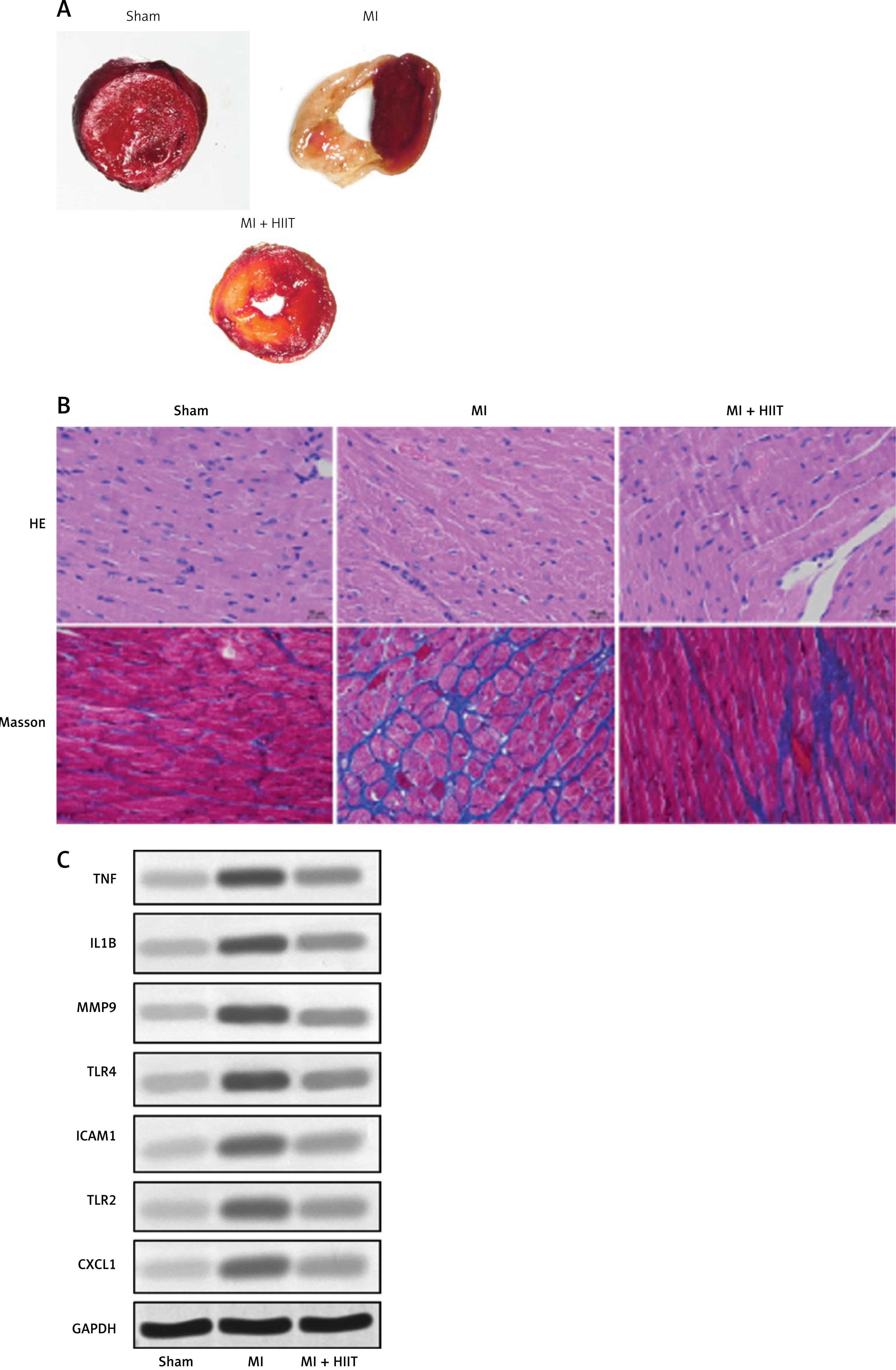
Histological analysis further supported these findings. Hematoxylin and eosin (HE) staining demonstrated disrupted myocardial architecture and increased cellular infiltration in the MI group, while the MI + HIIT group exhibited more preserved tissue structure and reduced inflammatory cell presence. Masson’s trichrome staining showed prominent collagen deposition (blue staining) in the MI group, which was significantly alleviated in the MI + HIIT group, indicating attenuated fibrosis (p < 0.05; Figure 7 B).
Western blot analysis of heart tissue confirmed that HIIT intervention substantially reduced the expression of all seven previously identified hub proteins – TNF, IL1B, MMP9, TLR4, ICAM1, TLR2, and CXCL1 – compared to the untreated MI group (Figure 7 C). GAPDH was used as the loading control. These findings provide strong experimental evidence that HIIT mitigates MI-induced damage by downregulating key inflammatory and fibrotic mediators in cardiac tissue, consistent with our bioinformatic predictions.
Discussion
Exercise remains a foundational pillar in cardiac rehabilitation (CR) [18], endorsed by global guidelines for the management of coronary heart disease (CHD) [19]. Research indicates that exercise-based CR significantly reduces the incidence and mortality associated with myocardial infarction, prevents cardiac remodeling, and enhances both the quality of life and functional capacity of patients after myocardial infarction [20]. HIIT is increasingly recognized as a safe and effective method for improving cardiac function in patients with myocardial infarction. The present study identified seven targets associated with HIIT in the context of myocardial infarction through bioinformatics analysis. It has been demonstrated that these key targets are modulated by HIIT in rat models of myocardial infarction.
While HIIT offers potent benefits, it may not be feasible for all patients, especially those recovering from AMI who face limitations due to fatigue, comorbidities, or risk of overexertion. In such cases, regular physical exercise in general – including moderate-intensity activities such as daily walking or low-impact aerobic routines – plays a crucial role in cardiac rehabilitation. Evidence from meta-analyses shows that even simple increases in daily step count are associated with reduced all-cause and cardiovascular mortality, making it a practical option for broader patient populations [21]. This underscores the value of tailored exercise prescriptions that prioritize safety and sustainability over intensity alone, complementing more vigorous protocols such as HIIT where appropriate.
With the advance of computer technology, bioinformatics analysis has shown great potential in exploring disease pattern and providing promising diagnostic biomarkers for MI [22, 23]. A key limitation of this study is the use of different cell types across analyses, which may affect the joint interpretation of results. Additionally, the relatively small sample sizes in the human (n = 20 per group) and animal (n = 8 per group) validation studies may limit the statistical power to detect smaller effect sizes and the generalizability of the findings. The absence of multivariate analysis, due to the homogeneity of the rat cohort (same age and sex) and the acute timing of human sample collection (within 12 h of MI onset), may have missed potential confounding factors such as comorbidities or treatment effects. Future studies should incorporate multivariate analyses to account for such variables and validate these findings in larger, more diverse cohorts, including human cardiac tissue, to enhance translational relevance. The GSE66360 dataset profiles mRNA expression in circulating endothelial cells, which are relevant to systemic inflammation and vascular responses in MI but may not fully reflect changes in cardiac tissue. In contrast, our experimental validation used peripheral blood samples from patients (which include a mix of cell types) and cardiac tissue from rats, where HIIT directly impacts local inflammation and remodeling. While circulating endothelial cells provide insight into systemic inflammatory changes after MI, their gene expression profiles may differ from those in cardiac tissue or other cell types involved in MI pathology, such as cardiomyocytes or infiltrating immune cells. This discrepancy could influence the generalizability of our findings, particularly regarding the direct effects of HIIT on the myocardium. Future studies should aim to validate these hub genes in matched cell types, such as cardiac tissue from both human and animal models, to strengthen the translational relevance of the findings.
HIIT refers to repeated bouts of short-to-moderate-duration high-intensity exercise, interspersed with periods of low-intensity exercise, and has been shown to be more effective in improving VO2 peak, exercise capacity, and activities of daily living [24, 25]. Some previous studies have explored the potential targets involved in HIIT-mediated cardiac protection from MI. For example, Heiat et al. reported that HIIT can induce the upregulation of PGC-1α, TFAm, and VEGF levels in MI rats to increase mitochondrial biogenesis and angiogenesis [26]. Lu et al. found that HIIT alleviates oxidative stress in MI by reducing MDA levels and elevating SOD and GPx levels in rats. Additionally, HIIT was demonstrated to inactivate the PI3K/Akt pathway and activate the p38 and AMPK pathways in MI [27]. Nori et al. reported that HIIT significantly reduced the levels of KYN, MDA, Cyp1a1, and Ido1 in MI rats and more effectively inhibited the Ido1-Kyn-Ahr axis compared with moderate-intensity continuous training [28]. In our study, we discovered 481 DEGs when comparing MI with the control group in the GSE66360 dataset. GSEA showed that the DEGs were enriched in TLR and MAPK pathways. After intersecting with HIIT-related genes, we identified 39 HIIT-related DEGs in the MI group.
GO enrichment analysis revealed that these 39 targets were linked to the regulation of nitric oxide (NO) biosynthetic and metabolic processes. Studies have shown that nitric oxide critically affects exercise performance [29, 30]. Improving the bioavailability of NO is a potential strategy for improving exercise ability and cardiovascular health [30–32]. KEGG enrichment analysis revealed that these 39 targets were enriched in the TNF pathway, cytokine-cytokine receptor interaction, NF-κB pathway, IL-17, AGE-RAGE, and Toll-like receptor signaling pathways. Previous research has shown the critical involvement of the mentioned pathways in MI progression [33, 34]. A bioinformatics analysis study also revealed that DEGs in MI were enriched in IL17 signaling, TNF signaling, Toll-like receptor signaling, and MAPK pathways based on functional enrichment analysis, which was consistent with our results [35]. Our results suggest that HIIT may improve heart function in MI patients by targeting these associated signaling pathways.
A PPI network was constructed to identify key HIIT-related targets in MI. We screened 7 key genes using the NetworkAnalyzer function and the CytoHubba plugin in Cytoscape software, including TNF, IL1B, MMP9, TLR4, ICAM1, TLR2, and CXCL1. Studies have reported that TNF is elevated following MI and is a potential prediction factor for infarction area [36]. Targeting TNF has been shown to alleviate the inflammation and injury in MI animal models [37–39]. HIIT has also been demonstrated to downregulate TNF levels in various diseases [40, 41]. IL1B is the main product of the active inflammasome and is elevated following MI [42]. Depletion of IL-1 reduces apoptosis and relieves myocardial remodeling in MI [43]. MMP9 is critical for post-MI left ventricle remodeling [44], and regulation of MMP contributes to improved heart function of MI rats [45]. TLR4 plays a critical role in the inflammasome formation in the heart. HIIT is reported to inhibit the NLRP3 inflammasome activation and relieves MI injury by reducing TLR4, IL1B, and MMP9 levels [46]. ICAM1 is a member of the immunoglobulin superfamily of cell adhesion molecules and is involved in various cardiovascular diseases [47]. ICAM1 expression is at a high level in MI patients and associated with major adverse cardiac events [48]. HIIT has been demonstrated to reduce the levels of TNF and ICAM1 in the serum of chronic heart failure patients [49]. TLR2 is increased after MI, and inhibition of TLR2 reduces infarct size in MI mice [50]. Knockdown of TLR2 can downregulate IL-6 and ICAM1 levels, reduce inflammatory response, and decrease infarct size in MI mice [51]. CXCL1 has also been identified as an upregulated gene and an independent risk factor in MI in previous studies [35, 52]. Exercise was demonstrated to promote CXCL1 production in mouse serum [53]. Consistently, in our study, we investigated the differential expression for these 7 targets in the GSE66360 dataset. We found that they were all upregulated in the MI group. Further validation using RT-qPCR identified their upregulation in patients with MI compared with healthy individuals, which was in line with previous findings. Moreover, in MI rat models, HIIT reduced infarct size and significantly downregulated the expression of seven key targets, suggesting their involvement in HIIT-mediated protection against MI.
Beyond exercise interventions, preventing CVD events and mortality in post-AMI patients requires a comprehensive, multifaceted approach that integrates lifestyle changes, risk factor management, and evidence-based therapies. This includes smoking cessation, dietary improvements, blood pressure control, lipid management, and adherence to medications, all tailored to individual patient profiles. Such holistic strategies, as outlined in expert consensus guidelines, can significantly enhance long-term outcomes by addressing the multifactorial nature of cardiovascular risk [54]. By combining these elements with exercise, clinicians can optimize secondary prevention and reduce recurrent events in this high-risk group.
Additionally, this study also possesses some other limitations. First, the sample size of the validation study in human subjects and rats was relatively small, which might affect the generalization of the results. Future research is warranted to test the performance of key biomarkers in large sample size and multiple centered studies. Second, the regulatory mechanisms related to the hub genes in MI progression were not explored in this study. Future studies should explore the upstream and downstream regulatory mechanisms of selected hub genes to elucidate their explicit role in MI. Third, the dynamic expression changes of different biomarkers were not monitored during the animal study, and the association between gene expression and exercise intensity/duration was not fully determined. Despite these limitations, this study identified seven HIIT-related key targets in MI (TNF, IL1B, MMP9, TLR4, ICAM1, TLR2, and CXCL1) based on bioinformatics analysis, and these seven biomarkers were found to be upregulated in MI patients and rat models. The explicit mechanism by which HIIT regulates these 7 key genes in MI requires further investigation. The findings of our study may deepen our understanding of the molecular basis of HIIT in MI therapy and provide novel insights into cardiovascular health management.
In conclusion, this study suggests that high-intensity interval training (HIIT) may contribute to cardioprotection in myocardial infarction by downregulating seven key inflammatory genes – TNF, IL1B, MMP9, TLR4, ICAM1, TLR2, and CXCL1. These molecular changes were associated with reduced infarct size and tissue remodeling in a rat model, highlighting HIIT’s potential as a therapeutic strategy. However, the preliminary nature of these findings, due to the small sample size and lack of multivariate analysis, warrants further validation in larger studies to establish clinical relevance.
By bridging bioinformatics predictions with in vivo validation, our findings highlight how exercise can modulate critical pathological pathways at the molecular level. While the use of different tissue types across analyses is a limitation, the consistency of gene regulation across models supports the robustness of our conclusions. Future work should focus on validating these targets in human cardiac tissue and exploring patient-specific HIIT protocols for optimized care.