Introduction
Systemic lupus erythematosus (SLE), a kind of severe autoimmune disease (AID), is characterized by loss of immunological tolerance to self-nuclear antigens, abnormal T- and B-cell responses and autoantibody production [1, 2]. To date, the pathogenesis of SLE remains uncertain [3]. Particularly much remains to be illuminated about gene expression levels and related regulatory mechanisms in SLE. Nowadays, emergence of transcriptomics shift the trend toward genome-wide expression, and becomes an important method to identify the gene signatures and biological function. In the past, extensive studies about transcriptomics mainly paid attention to protein-coding genes. Recently, studies about transcriptomics began to focus on noncoding RNA (ncRNA) transcripts. ncRNAs are RNA molecules which are not translated into proteins [4]. Long ncRNA (lncRNA) and micro RNA (miRNA) are two major types of ncRNAs, and as crucial regulators can be found in various biological processes by interfering with gene expression [5].
LncRNA is larger than 200 nt in length. Recently, many studies have demonstrated the important role of lncRNAs in the immune system, including dendritic cell differentiation, T cell activation and granulocytic differentiation, etc. [6, 7]. But studies on the role of lncRNAs in SLE are very limited. Growth arrest specific 5 (GAS5) was reported to be connected with increased susceptibility to SLE in a mouse model; linc0597 and linc0949 were found significantly decreased in SLE patients [8–10]. In our recent study, linc-DC and GAS5 were decreased and linc0597 overexpressed in plasma of SLE patients, and linc0597, GAS5 and lnc-DC can be used as potential biomarkers for SLE [11]. miRNAs range from 18 to 25 nt in length and regulate approximately 90% of protein-coding genes [12]. In previous studies, many miRNAs such as miR-155, miR-17, miR-181b, miR-142-3p and miR-326 were reported to be related to the pathogenesis of SLE [13–15]. However, there are no studies investigating mRNAs, lncRNAs and miRNAs expression simultaneously in SLE.
As a revolutionary tool for transcriptomic study, RNA sequencing (RNA-seq), the first sequencing-based method, can act as a high-throughput and quantitative survey of the complete set of transcripts; therefore it has become an important and widely used method for transcriptomic research [16, 17]. Recently, RNA-seq has also been used in several studies of SLE pathogenesis [18, 19].
In the present study, to identify the integrated expression profile of lncRNA, miRNA and mRNA in SLE, RNA-seq was performed. In addition, to describe the underling biological functions of differentially expressed RNAs, bioinformation analysis was conducted.
MATERIAL AND METHODS
Subjects
All participants in our study were in accordance with the ethical standards of the institutional and/ or national research committee, as well as the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent from all participants in our study was also collected. The SLE patients were recruited from Anhui Provincial Hospital and the First Affiliated Hospital of Anhui Medical University. The age- and sex-matched healthy controls (HC) were recruited from the Physical Examination Center of the First Affiliated Hospital of Anhui Medical University. Finally our study included 24 SLE patients and 24 HC. The inclusion criteria were according to the SLE classification of the American College of Rheumatology in 1997; all patients must fulfill at least four of the criteria [20]. A questionnaire survey was used to collect demographic data and clinical manifestations of all patients. Laboratory data were obtained from the medical record. The SLE disease activity index (SLEDAI) was used to evaluate disease activity of SLE patients [21]. Active SLE was defined as a SLEDAI score ≥ 8. Clinical manifestations and laboratory records of all subjects are shown in Table I.
Table I
Demographics, clinical manifestations and laboratory characteristics of subjects
Blood samples
Blood samples (5 ml) were harvested directly from all participants. All blood samples were drawn by venepuncture and collected into PAXgene Blood RNA tubes for transcriptomics assays, and allowed to stand at room temperature for 2 h, then frozen at –20°C overnight, and then stored at –80°C. Equal amounts of sample from 8 different individuals (SLE patients and HC group) were randomly pooled to minimize the biological variation. In total, there were three samples in the SLE group, and three in the HC group.
Transcriptomics analysis of blood samples (RNA sequencing, RNA-seq)
mRNA Prior to library preparation, the concentration and integrity were detected for each sample by an Agilent 2,100 Bioanalyzer (Agilent RNA 6000 Nano Kit).
Firstly, total RNA was extracted from mixed samples of with equal weights. Subsequently, Oligo-dT beads were used to purify the total RNA sample (200 ng), then poly (A)-containing mRNA was fragmented into small pieces with elute, prime and fragment mix. First strand cDNA was then synthesized via the cleavage of short mRNA fragments. Then the second-strand cDNA was synthesized, and the cDNA fragments were subjected to end repair. After end-repair and adapter-ligation, the products were amplified through PCR and purified to construct the cDNA library.
The library preparations were sequenced on an Illumina Hiseq 2000 platform; read length 90 nt is the most common sequencing strategy.
LncRNA Ribo-Zero Magnetic Kit (Human/Mouse/Rat) (Epicentre) was used to deplete rRNA from 5 μg of total RNA. The retrieved RNA was fragmented by adding First Strand Master Mix. The library preparations were the same as mRNA.
miRNA Approximately 1 μg RNA from each sample was used to construct the subsequent cDNA library following the protocol of TruSeq Small RNA Sample Prep Kits. RNAs were ligated to 3′ and 5′ adaptor sequentially, reverse transcribed to cDNA and PCR amplified. Then the PCR products were purified with PAGE gel after ethanol precipitation and washing, and then the purified small RNA libraries were quantified and used for cluster generation and 36 nt single end library preparations were sequenced on the Illumina Hiseq 2000 platform.
Statistical analysis
Data were expressed as (x ± s) or medians with range. Comparison of continuous data was performed by independent Student’s t-test. The Statistical Package for the Social Sciences (SPSS) 23.0 for Windows (IBM Co., Armonk, NY, USA) was used for statistical analyses. P-values < 0.05 were considered statistically significant.
Sequencing data were analyzed by using fold changes (FC) and Student’s t-test. lg|FC| ≥ 2.0 and Padj ≤ 0.05 were set as the criterion to identify the differentially expressed mRNAs and lncRNAs. And lg|FC| ≥ 1.0 and Padj ≤ 0.3 were set as the threshold to identify the aberrantly expressed miRNAs.
Functional group analysis
The filtered co-expressed genes were defined as potential target genes of lncRNAs. Pearson and Spearman correlations were calculated for each pair of genes and the correlation coefficient ≥ 0.6 as our criterion.
We used the miRTarBase to predict the potential targets of those differentially expressed miRNAs miRTarBase (http://mirtarbase.mbc.nctu.edu.tw) is a database that provides the latest and extensive validated miRNA-target interaction information [22].
To determine the division of SLE relevant and irrelevant RNAs, we conducted the Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways analysis.
GO (http://www.geneontology.org) analysis was used to detect the function of that associates distinctively expressed genes with GO terms and reveals gene regulatory networks on the basis of cellular components, biological processes and molecular functions [23, 24]. The hypergeometric test was used to get the target GO terms. If the p-value is less than 0.05, the GO term is significant enrichment in differentially expressed RNAs. KEGG (http://www.genome.jp/kegg/) analysis is used to identify the pathways of these differentially expressed RNAs enriched by Fisher’s exact test [25, 26]. The KEGG analysis was the same as GO enrichment. If the Padj is less than 0.05, the pathway is significant enrichment in differentially expressed RNAs.
RESULTS
Summary of RNA-seq
Deep sequencing generated the mean number of raw reads, 273,119,891.3 and 266,323,616.7, for SLE patients and the HC group, respectively. Removing reads with non-canonical letters or with low quality, and discarding the sequences shorter than 18 nt, the mean numbers of clean reads were 259,455,621.3 and 252,555,396.0, for SLE patients and the HC group, respectively, which remained for analysis (Table II). The filtered clean reads has been submitted to the American National Center for Biotechnology Information (Num: SRP076773). A total of 93,267 transcripts were detected in the current study, in which 37,637 transcripts were identified as mRNA and 55,630 transcripts as lncRNA.
Table II
Sequence comparison results of RNA-seq
Differentially expressed lncRNAs and mRNAs
In total 2,354 lncRNAs and 827 mRNAs were identified to be significantly expressed. Among them, 1,873 known lncRNAs (221 up-regulated and 1,652 down-regulated) were identified in SLE patients. Additionally, 481 novel lncRNAs (73 increased and 408 decreased) were revealed (Supplementary Tables SI, SII). We also found that a total of 78 known mRNAs and 2 novel mRNAs increased, and 729 known mRNAs and 18 novel mRNAs decreased in the SLE patients (Supplementary Tables SIII, SIV, Figures 1, 2).
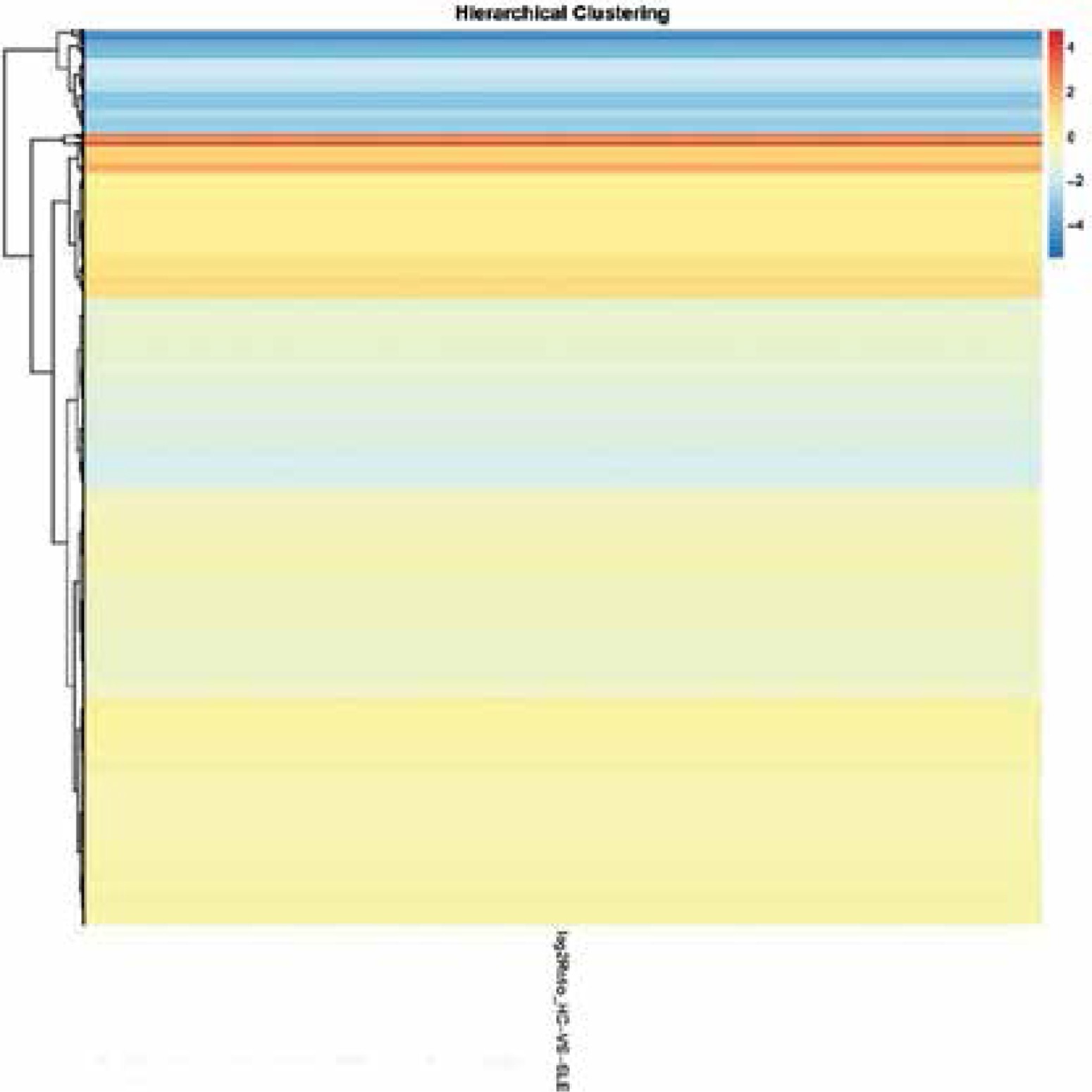
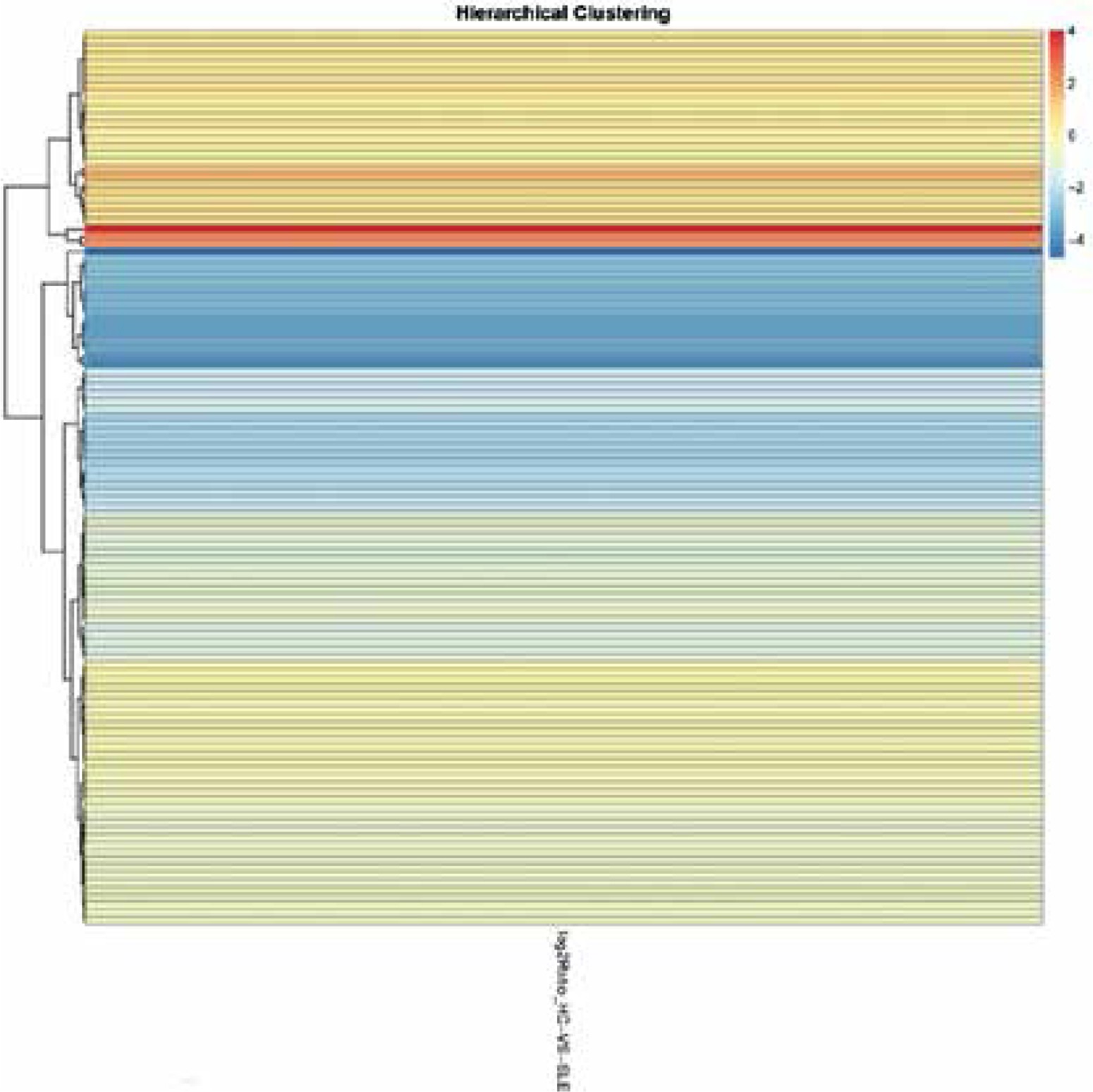
mRNAs GO and KEGG analyses
GO analysis was used to explore the function of the significantly altered genes (Supplementary Table SV). The differentially expressed mRNAs were mainly enriched in nine molecular function terms containing ribonucleotide, purine nucleotide, and protein serine/threonine kinase activity, and one biological process term of a cellular metabolic process.
The KEGG pathway analysis was performed to identify pathways which were significantly enriched with differentially expressed RNAs (p < 0.05). In total, differentially expressed mRNAs significantly enriched 16 pathways (Supplementary Table SVI). Among these pathways, the organismal systems pathways category contained the largest number of differentially expressed genes and the FcγR-mediated phagocytosis pathway contained the most differentially expressed genes.
GO and pathway analysis of differentially expressed lncRNA-targeted mRNA
Since the function of lncRNAs is mainly executed in protein-coding target genes, we utilized the co-expression patterns of mRNAs and lncRNAs to infer biological functions for the latter. These genes were considered as potential target genes of lncRNAs. We then used GO and KEGG analyses to identify the inferred functions about the differentially expressed lncRNAs’ target genes between SLE patients and the HC group. At last, ten GO terms in cell component and five in molecular function were significantly enriched. These five GO terms in molecular function were ion binding, cation binding, metal ion binding, transition metal ion binding and zinc ion binding (Supplementary Table SVII).
The pathway analysis of lncRNAs was also conducted. These differentially expressed lncRNA-targeted mRNAs were significantly enriched in ten pathways in our study (Supplementary Table SVIII). Among them, the environmental information processing pathways category contained the largest number of differentially expressed genes, which fell into two major subgroups involved in the phospholipase D signaling pathway and cAMP signaling pathway. And the results of KEGG pathway analysis of mRNA showed that the FcγR-mediated phagocytosis pathway, glycosaminoglycan biosynthesis-chondroitin sulfate/dermatan sulfate and glyoxylate and dicarboxylate metabolism were also significantly enriched.
miRNA-seq summary
The mean numbers of raw reads were generated from the deep sequencing of the small RNA libraries, 11,925,626 and 11,933,904, for SLE patients and the HC group, respectively. The mean number of clean reads, 11,548,206 and 11,518,853, for SLE patients and the HC group, respectively, remained for analysis (Table III). Length distribution of sequenced miRNAs ranges from 15 to 30 nt, with a sharp peak at 20–24 nt.
Table III
Sequence comparison results miRNA-seq
Differentially expressed miRNA
In the miRNA sequencing study, 24 miRNAs were differentially expressed. Among these miRNAs, there were 6 increased and 18 decreased miRNAs in SLE patients (Supplementary Table SIX). hsa-miR-144-5p, hsa-miR-1304-3p and hsa-miR-30b-3p were the most significantly down-regulated miRNAs, while hsa-miR-125b-5p, hsa-miR-1226-3p and hsa-miR-618 were the most significantly up-regulated miRNAs in the SLE group.
GO and pathway analysis of differentially expressed miRNA-targeted mRNA
To provide clues for the possible roles of 24 differentially expressed miRNAs, miRTarBase were used to conduct target prediction analysis.
First we performed GO analysis based on the set of target genes (Supplementary Tables SX, SXI). The results of GO analysis suggested that the main molecular function of up-regulated miRNAs was ankyrin binding; there were no enriched items in cell component and biological process. The main biological processes of down-regulated miRNAs involvement were cell differentiation, proliferation and activation; these cells included B cells, leukocytes, lymphocytes and mononuclear cells. All these cells play important roles in the immune system. Also one GO term in cell component and three in molecular function were significantly enriched.
Then the target predicted genes were grouped into different pathways. The up-regulated miRNAs were grouped into 52 pathways and 4 pathways were significantly enriched (Supplementary Table SXII). Among these pathways, the NF-κB signaling pathway played an important role in the immune system. The down-regulated miRNAs were grouped into 30 pathways and 3 pathways were significantly enriched, including metabolism of xenobiotics by cytochrome P450, bile secretion and terpenoid backbone biosynthesis pathways (Supplementary Table SXIII).
Discussion
Genome-wide association studies (GWAS) have shown some genetic risk factors for AIDs, and several genes may suggest common mechanisms that lead to the development and progression of AIDs, including SLE [27, 28]. In the past many studies have focused on RNAs, and aberrant expression of mRNA levels has been observed in SLE patients, such as IL-28RA, DNMT1 and DNMT3A [29, 30]. Recently, studies showed that ncRNAs can serve as important modulators in the development of AIDs [31]. However, the role of ncRNAs in the pathogenesis and development of SLE is still less understood. In the present study according to comparative analyses of the RNA-seq data, a total of 78 protein-coding RNAs were increased while 729 were decreased. Also, a total of 1,873 known lncRNAs (221 up-regulated and 1,652 down-regulated) and 481 novel lncRNAs (73 increased and 408 decreased) were significantly altered in SLE patients. Compared with a previous study, numerous differentially expressed RNAs in our study have been shown to play important roles in the pathogenesis of SLE, including receptor of IL-21 (IL-21R) [32], receptor of IL-23 (IL-23R) [33] and toll-like receptors 2 (TLR2) [34], etc. In addition, several differentially expressed genes such as Yippee-like-4 (YPEL4), Ubiquilin-4 (UBQLN4), interferon regulatory factor-6 (IRF6) and other genes were reported first in our study.
Previous studies have reported that miRNAs were critical for not only the development of the immune system, but also for the function of both innate and adaptive arms [35]. We found that 6 miRNAs were increased and 18 miRNAs were decreased. Among these differentially expressed miRNAs, hsa-let-7a-5p [36], hsa-miR-125b [37] and hsa-miR-148a [38] have been reported to connect with SLE. In addition, we identified several differentially expressed miRNAs that have not been reported previously in SLE, such as hsa-miR-144-5p, hsa-miR-1304-3p, hsa-miR-30b-3p.
Precisely annotating the functions of RNAs remains complex. In this study, according to the results of GO analysis, differentially expressed mRNAs were mainly involved in the molecular function terms of ribonucleotide, purine nucleotide, and protein serine/threonine kinase activity. Protein serine/threonine kinase activity is a major component of apoptotic factor. A previous study has shown that serine/threonine-protein kinase 17A (SKT17A) was associated with SLE susceptibility [39]. LncRNAs cover nearly all aspects of gene expression and protein translation. The main function of up-regulated miRNAs was ankyrin binding. The main biological process of down-regulated miRNA involvement was cell differentiation, proliferation and activation; these cells include B cells, leukocytes, lymphocytes and mononuclear cells. Especially the critical role of B-cells in the pathogenesis of SLE is well recognized – from producing autoantibody to abnormal regulation of the immune system [40].
Differentially expressed mRNAs and lncRNAs were both enriched in FcγR-mediated phagocytosis, glycosaminoglycan biosynthesis-chondroitin sulfate/dermatan sulfate and glyoxylate and dicarboxylate metabolism pathways. The FcγR-mediated phagocytosis pathway belongs to the immune pathways and contained the most differentially expressed genes, such as receptor for immunoglobulin G (FCGR)-1B and YPEL4. FCGRs play an important role in linking the humoral and cellular portions of the immune system [41]. When bound and clustered by immune complexes, FCGRs initiate a number of cellular processes aimed at the elimination of antigen. The FCGR gene family (FCGR2A, FCGR2B, and FCGR2C) was considered to be associated with the susceptibility to SLE [42]. YPEL4 is one of the important molecules in regulating cell proliferation [43]. However, currently, the molecular mechanisms of YPEL4 in SLE remain unclear. Previous studies showed that the pathway is associated with some other AIDs, such as RA and neuromyelitis optica [44, 45], but did not find the FcγR-mediated phagocytosis pathway in SLE.
The results of miRNA KEGG analysis showed that some up-regulated miRNAs were significantly enriched in the NF-κB signaling pathway, which plays an important role in the immune system. NF-κB is a kind of transcription factors which can regulate genes involved in inflammation, cell survival and immunity, and our previous study supported the important role of the NF-κB signaling pathway in the genetic basis of SLE [46]. Our results may contribute to future genetic studies in SLE pathogenesis and may promote the development of new therapeutic strategies by targeting these pathways.
Several limitations of our study should be acknowledged. First, different treatment strategies in the study subjects may influence the results of RNA expression levels. Second, further validation of those differentially expressed mRNAs, lncRNAs and miRNAs in a larger cohort is still needed to confirm our preliminary results.
In conclusion, the findings of this study demonstrate a comprehensive expression profile of lncRNAs in SLE with concurrent integrated expression of miRNAs and mRNAs and imply potential regulatory functions of lncRNAs, miRNAs and mRNAs which are implicated in the development and pathogenesis of SLE.