Introduction
Alzheimer’s disease (AD) is acknowledged as a progressive multifarious neurodegenerative disorder and the most common form of dementia in late adult life [1, 2]. AD is pathologically characterized by progressive neuronal degeneration and extracellular amyloidal protein deposits (senile plaques) [3, 4]. Oxidative stress (OS) has been demonstrated to be involved in the pathogenesis of AD and H2O2-induced oxidative damage is a typical early feature of AD [5]. Although the neuropathological features of AD have been identified, its pathogenesis has not been clearly revealed. Thus, there is an urgent need to identify the pathogenesis of AD, expecting to provide new potential therapeutic targets.
MicroRNAs (miRNAs) are endogenous, small, non-coding RNAs that participate in numerous biological processes by regulating the expression of their target genes [6–8]. Research into AD has revealed that miRNAs were involved in the cellular changes and interfere with gene regulation and translation [9]. In recent years, several studies have reported the altered expression of several miRNAs, such as miR-27a, miR-132 miR-29a, miR-29b, miR-125b and miRNA family let-7 in cerebrospinal fluid (CSF) of AD patients [10–14]. Furthermore, it has also been verified that miRNAs play a critical function in the progression of several pathologies, including AD, Parkinson’s disease (PD) and other neurodegenerative diseases [15, 16].
Among them, miRNA let-7 family members are key regulators of cell proliferation and developmental timing and have also been demonstrated to inhibit the tumor disease progression by regulating apoptotic genes and cell growth [17]. Notably, miR-let-7f, a member of the let-7 family, has been reported to be up-regulated in AD patients [18]. Additionally, miR-let-7f also exhibited an anti-apoptotic role and a protective effect on cell survival in AD models [19]. Given the above evidence, we speculated that miR-let-7f may participate in the pathogenesis of AD. However, the molecular mechanism of miR-let-7f in AD pathogenesis has not yet been determined.
Therefore, the present study was aimed to elucidate the underlying role of miR-let-7f in oxidative damage in SH-SY5Y cells, expecting to present a molecular mechanism of miR-let-7f in AD pathogenesis.
Material and methods
Human subjects
Blood samples were collected from patients in our hospital. The protocol for this investigation was approved by the Ethics Committee of our Hospital. Written informed consent was obtained from each participant. A total of 30 patients diagnosed with AD (aged 58 to 68 years) and 30 age-matched healthy volunteers (controls) were included in the study. The Mini-Mental State Examination (MMSE) was performed in all subjects [20]. All AD subjects underwent formal neuropsychological testing as previously described [21]. The age-matched healthy volunteers were recruited from normal elderly persons, who underwent medical screening carried out by a gerontologist to check whether the elderly persons were healthy. No healthy volunteer had a family history of AD. In addition, subjects who had a history of organic mental disorders, substance use disorders, major depression and bipolar disorder were excluded from healthy volunteers. The healthy volunteers were free of psychotropic medications. Subjects (either AD patients or healthy volunteers) with chronic metabolic and inflammatory conditions or acute illness were excluded from the study.
miRNA microarray
Blood samples from the median cubital vein of subjects were collected for miRNA microarray analysis. Blood samples for all subjects were collected and total RNA was isolated with TRIzol reagent (Invitrogen, USA) according to the manufacturer’s instruction. The miRNA was labeled by mirVana microRNA Labeling Kit (Ambion Inc. Austin, TX) according to the manual. Monoreactive Cy5 dye (Amersham Pharmacia Biotech, Ltd, Uppsala, Sweden) was used for dyeing. The fluorescent probes were lyophilized, and then re-suspended in 15 l of diethyl pyrocarbonate (DEPC) water and 5 l of 4 hybridization buffer, denatured by heating for 5 min at 65μC and then snap-cooled on ice for 15 min. The hybridization was carried out for 20 h at 42μC in a rotating hybridization oven. After hybridization, slides were washed and then scanned by a Generation III array scanner (Amersham Pharmacia Biotech, Ltd, Uppsala, Sweden). Hybridization was carried out twice on two different days.
Cell culture and oxidative stress
The human neuroblastoma cell line SHSY-5Y was originally obtained from PhD Hong Zhang, Department of Laboratory Center, Chinese Medical University (Shenyang, China). SHSY-5Y cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS, Invitrogen, USA) in an incubator at 37μC with a 5% CO2 atmosphere. Cells were subcultured when confluent (> 80% confluence) using 2.5 g/l trypsin. In order to induce the oxidative damage, SHSY-5Y cells (80% confluence) were exposed to 200 µM H2O2 for 24 h. Then cells were harvested to perform the cell viability assay, miR-let-7f expression measurement and cell morphological observation. The cell morphological was observed using a light microscope (magnification: 40×).
Cell viability assay and apoptosis assay
Cell viability was determined by cell counting Kit-8 (Beyotime, China). In brief, 5 × 103 cells were seeded into each well of 96-well plates, followed by incubation overnight at 37μC with a 5% CO2 atmosphere. Then, cells were treated with H2O2 according to the study design. Subsequently, the culture medium was replaced by basal medium containing 10% CCK-8 solution, and the incubation continued for another 1 h at 37μC. Absorbance was measured at 450 nm by a microplate reader (Bio-Rad, USA).
Cell apoptosis was analyzed with flow cytometry assay. Apoptosis was detected using the FITC Annexin V/Dead Cell Apoptosis Ki (Invitrogen, USA) according to the manufacturer’s protocol. Cells were stained with 5 µl of FITC-annexin V and 0.1 µg propidium iodide (PI), and then incubated at room temperature for 15 min. Apoptosis rate was measured through flow cytometry (MoFlo XDP, Beckman, USA). Percentage of apoptotic cells was analyzed by using Kaluza Analysis Software.
siRNA transfection
siRNA transfection was performed with Superfect Transfection Reagent (Qiagen, USA) according to the manufacturer’s instructions. The miR-let-7f mimics, miR-let-7f-1 inhibitors (anti-miR-let-7f) and their corresponding controls (scramble and negative controls, respectively) were chemically synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China). Cells were transfected with 50 nM oligonucleotides for 48 h and then harvested. The overexpression and inhibition of target proteins were confirmed by RT-PCR after transfection.
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s instructions. The cDNA of miR-let-7f and AKT-2 was respectively prepared using the One Step PrimeScript miRNA cDNA Synthesis Kit (TaKaRa, China) and PrimeScript RT Reagent Kit (TaKaRa, China). The qRT-PCR analysis was performed using the SYBR Green RT-PCR Kit (Takara Bio, Japan) on an ABI 7500 Fast Real-Time PCR System (Applied Biosystems, USA). The small nuclear RNA U6 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) respectively were used as the endogenous control of let-7f and AKT-2 to calculate the relative RNA levels. Data were calculated by the comparative cycle threshold (CT) (2– Δ Δ CT) method. The PCR primer sequences were as follows.
(1) miR-let-7f forward: 5’-CTATACAATCTATTGCCTTCCC-3’;
(2) U6, forward primer: 5’-TGCGGGTGCTCGCTTCGGCAGC-3’. The reverse primers for miR-let-7f and U6 were universal adaptor primers available in a ready-to-go format (One Step PrimeScript miRNA cDNA Synthesis Kit, D350A; Takara, China).
(3) AKT-2 forward: 5’-AGGCACGGGCTAAAGTGAC-3’; and reverse: 5’ CTGTGTGAGCGACTTCATCCT-3’.
(4) GAPDH forward: 5’-CTGGGCTACACTGAGCACC-3’; and reverse: 5’-AAGTGG TCGTTGAGGGCA ATG-3’.
Western blot
Whole-cell lysates were prepared using radioimmunoprecipitation assay (RIPA) lysis buffer (Thermo Fisher, USA). Protein concentration was measured using BioRad protein assay reagent (BioRad, USA). Equal amounts of proteins (40 µg) of each sample were separated by 10% sodium dodecyl sulfate-polyacrylimide gel electrophoresis (SDS–PAGE) and then transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, USA). PVDF membrane was blocked in skim milk at 25μC for 2 h, and subsequently incubated at 4μC overnight with antibodies against AKT-2, Bcl-2, Bax, active caspase-9, active caspase-3 or GAPDH (all in 1 : 1000 dilution, Santa Cruz, USA) in Tris-buffered saline with Tween-20 (TBST) containing 5% defatted milk. Next, PVDF membrane was incubated with corresponding horseradish peroxidase-conjugated (HRP)-linked secondary IgG antibodies (anti-mouse or anti-rabbit, 1 : 1000 dilution, Santa Cruz, USA) for 1 h at room temperature. The bands of bound secondary antibody were detected with an enhanced chemiluminescence kit (Pierce Biotechnology, USA) and the signals were detected with a SuperSignal Protein Detection kit (Pierce Biotechnology, USA). The band intensity of western blot was quantified subsequent to normalization with the density of GAPDH using ImageJ (National Institutes of Health, USA).
Luciferase reporter gene assay
For the luciferase reporter gene assay, SHSY-5Y cells were cultured in 96-well plates at 37μC for 24 h until 70–80% confluence. The 3’-UTR region of AKT-2 was cloned downstream of the Renilla luciferase stop codon in the psiCHECK vector (Promega, USA). Then, p3’-UTR-AKT2 or p3’-UTRmut-AKT2luciferase reporter vectors together with miR-let-7f mimics or miR-let-7f inhibitors were co-transfected into SHSY-5Y cells using Superfect Transfection Reagent (Qiagen, USA). After 48 h, the luciferase activity was determined by a dual-luciferase reporter assay system (Promega, USA). All transfection experiments were conducted in triplicate and repeated 3 times independently.
Statistical analysis
Statistical analysis was performed using SPSS software version 20.0 (SPSS, Inc., USA). Results were expressed as mean ± standard error of mean (SEM) of at least three separate experiments. Statistical significance was determined with one-way ANOVA or Student’s t-test. Differences between two groups was analyzed using Student’s t-test or χ2 test. All statistical tests were two-sided; significance was set at p < 0.05 along with 95% confidence intervals (CI).
Results
miRNA microarray analysis verified the up-regulation of miR-let-7f in AD patients
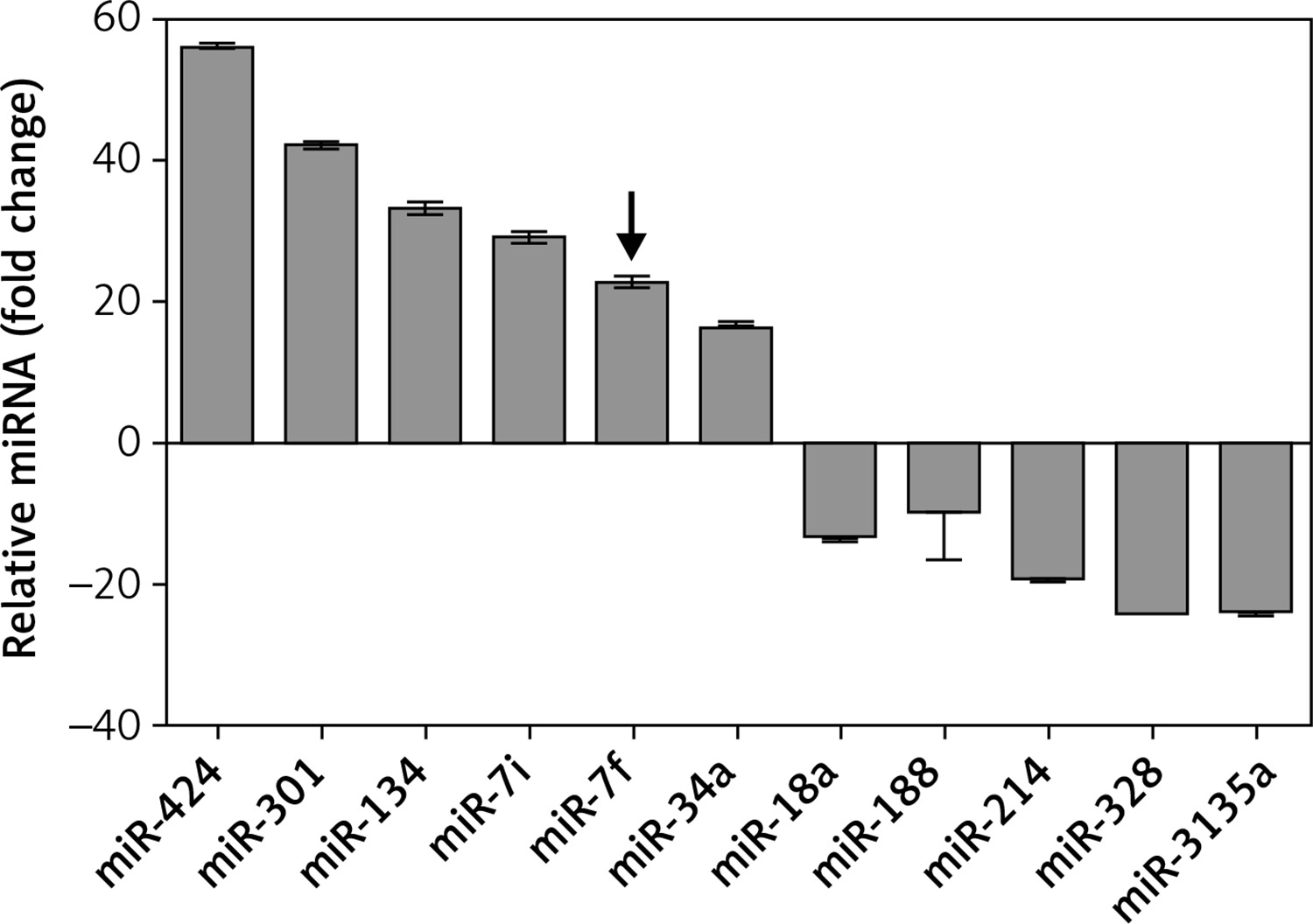
A total of 30 patients diagnosed with AD (aged 58 to 68 years) and 30 age-matched healthy volunteers (aged 55 to 70 years) were included in the study. The characteristics of the included subjects are listed in Table I. The two groups were well balanced in terms of gender (p = 0.34), age (p = 0.14), years of education (p = 0.75), and disease history (p > 0.05). The MMSE score of AD patients was significantly lower than that of healthy volunteers (p = 0.005). To explore the potential function of miRNAs in the pathogenesis of AD, miRNA microarray analysis was performed to detect the expression levels of various miRNAs. The results showed that 11 miRNAs were differentially expressed in the AD patients compared with controls (Figure 1). Among them, 6 miRNAs – miR-424, miR-301, miR-134, miR-let-7i, miR-let-7f and miR-34a – were significantly up-regulated and 5 miRNAs – miR-18a, miR-188, miR-214, miR-328 and miR-3135a – were significantly down-regulated. The miRNA microarray analysis verified that miR-let-7f was up-regulated in AD patients. Taken together, the results indicated that miR-let-7f was involved in the pathogenesis of AD.
Table I
Characteristics of the included subjects
miR-let-7f is involved in the H2O2-induced oxidative damage in SH-SY5Y cells
H2O2-induced oxidative damage is a typical early feature of AD. Thus, in order to simulate the oxidative damage of AD in vitro, SH-SY5Y cells were exposed to H2O2 to induce oxidative damage. Firstly, in order to optimize the dosage of H2O2, SH-SY5Y cells were incubated with 0, 50, 100, 200 or 500 µM H2O2, followed by assessments of cell viability. After H2O2 treatment, cell viability was significantly decreased compared to the untreated cells (p < 0.001, Figure 2 A). In subsequent experiments, the H2O2 concentration of 200 µM was used to induce oxidative damage.
Figure 2
H2O2-induced oxidative damage in SHSY5Y cells. A – Cells were exposed to H2O2 (0, 50, 100, 200 or 500 μM), and cell viability was measured by CCK-8 assay. B – Cells were treated with 200 μM H2O2, and non-treated cells acted as a control. Apoptosis rate of SH-SY5Y cells was tested by flow cytometry assay. C – The expression of apoptosis-related proteins was tested by Western blot analysis. D – The expression of miR-let-7f in SHSY5Y cells was measured by qRT-PCT
Data presented are the mean ± SEM (n = 3). **p < 0.01, ***p < 0.001 vs. control.
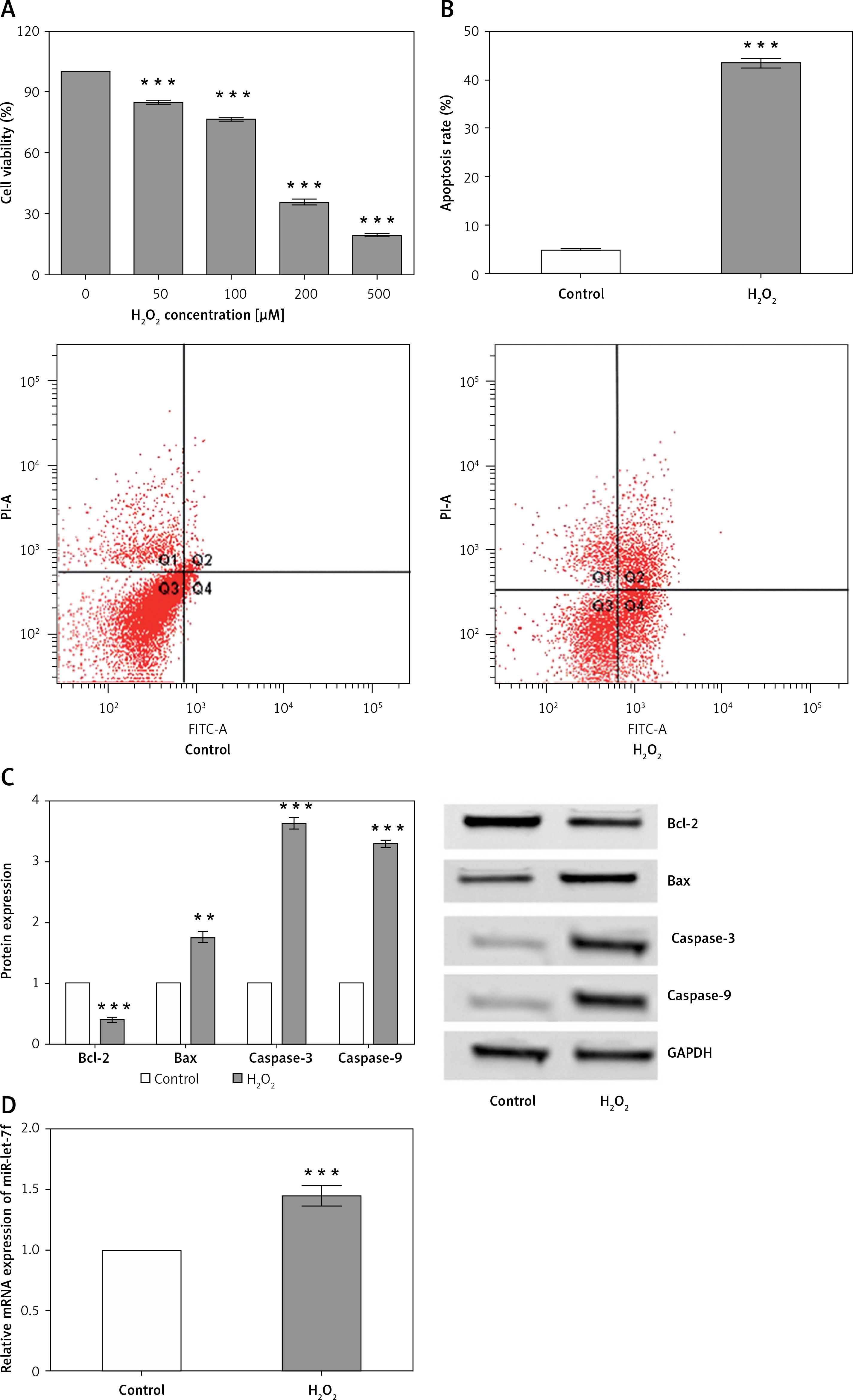
The apoptosis rate was examined by flow cytometry assay. After 200 µM of H2O2 treatment, the apoptosis rate of SH-SY5Y cells was significantly enhanced compared to the control (p < 0.001, Figure 2 B). Subsequently, we detected the expression of apoptosis-related proteins using western blot analysis. The results showed that anti-apoptotic Bcl-2 was significantly down-regulated after H2O2 treatment (p < 0.001), while the apoptosis-related proteins, including pro-apoptotic Bax (p = 0.001), cleaved caspase-3 (p < 0.001), and cleaved caspase-9 (p < 0.001), were significantly up-regulated (Figure 2 C). Taken together, the results indicated that H2O2 stimulation could reduce cell viability and promote apoptosis, thus inducing oxidative damage in SH-SY5Y cells.
Additionally, we detected the expression of miR-let-7f in H2O2-treated cells. Notably, we verified that miR-let-7f was significantly up-regulated in H2O2-treated cells compared with that in the control group (p = 0.0003, Figure 2 D). The results indicated that miR-let-7f was involved in the H2O2-induced oxidative damage in SH-SY5Y cells. The results of in vitro experiments were consistent with those of the clinical study, which further confirmed that miR-let-7f might participate in the pathogenesis of AD.
miR-let-7f up-regulation protected against H2O2-induced oxidative damage in SH-SY5Y cells
To further verify the function of miR-let-7f in the pathogenesis of AD in vitro, miR-let-7f mimic, miR-let-7f inhibitor (anti-miR-let-7f) and their corresponding controls, scramble (mimic NC) and negative controls (inhibitor NC) were transfected into SH-SY5Y cells, respectively, to alter the expression level of miR-let-7f. Then, the expression of miR-let-7f was confirmed by qRT-PCR (Figure 3 A). We observed that miR-let-7f was significantly up-regulated in SH-SY5Y cells transfected with miR-let-7f mimic (p < 0.001), while it was down-regulated in SH-SY5Y cells transfected with miR-let-7f inhibitor (p = 0.0004, Figure 3 A). These results showed that the changes in expression level of miR-let-7f were successfully achieved.
Figure 3
Effects of miR-let-7f on H2O2-induced oxidative damage in SH-SY5Y cells. A – SH-SY5Y cells were transfected with minic-NC, miR-let-7f mimic, inhibitor NC or miR-let-7f inhibitor, and non-transfected cells acted as a control. Expression of miR-let-7f was measured by qRT-PCR. B – Transfected and non-transfected cells were treated with 200 μM H2O2, and the cell viability was measured by CCK-8 assay. C – Transfected and non-transfected cells were treated with 200 μM H2O2, and the apoptosis rate was measured by flow cytometry assay. D – Transfected and non-transfected cells were treated with 200 μM H2O2, and the cell morphological was observed (magnification: 40 ×)
Data presented are the mean ± SEM (n = 3). ***p< 0.001.
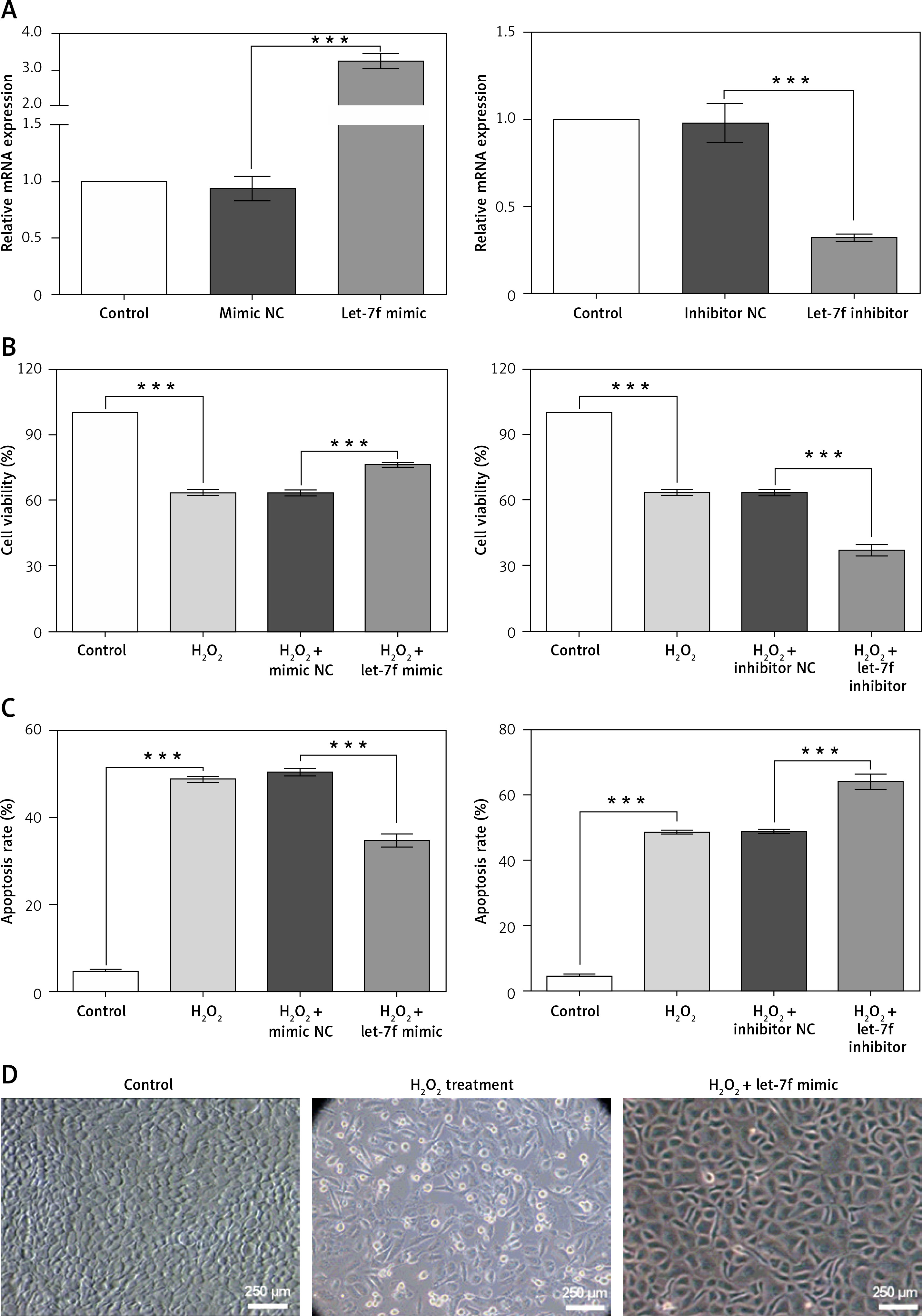
Then, the effects of miR-let-7f on the viability and apoptosis of H2O2-treated SH-SY5Y cells were evaluated. Even in the presence of H2O2, the up-regulation of miR-let-7f significantly increased the cell viability and inhibited cell apoptosis, compared to the negative controls (p < 0.001, Figures 3 B, C). However, the down-regulation of miR-let-7f significantly inhibited the cell viability and promoted cell apoptosis, compared to the negative controls (p < 0.001, Figures 3 B, C). In addition, we found that the H2O2 treatment caused cell morphological damage, while the up-regulation of miR-let-7f alleviated the H2O2-induced morphological damage (Figure 3 D). Overall, these results suggested that the up-regulation of miR-let-7f reversed the H2O2-induced decrease of cell activity, apoptosis and morphological damage, further indicating that miR-let-7f up-regulation protected against H2O2-induced oxidative damage in SH-SY5Y cells.
AKT-2 is the direct target of miRNA-let-7f
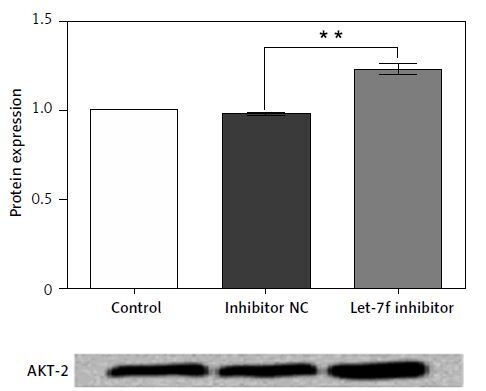
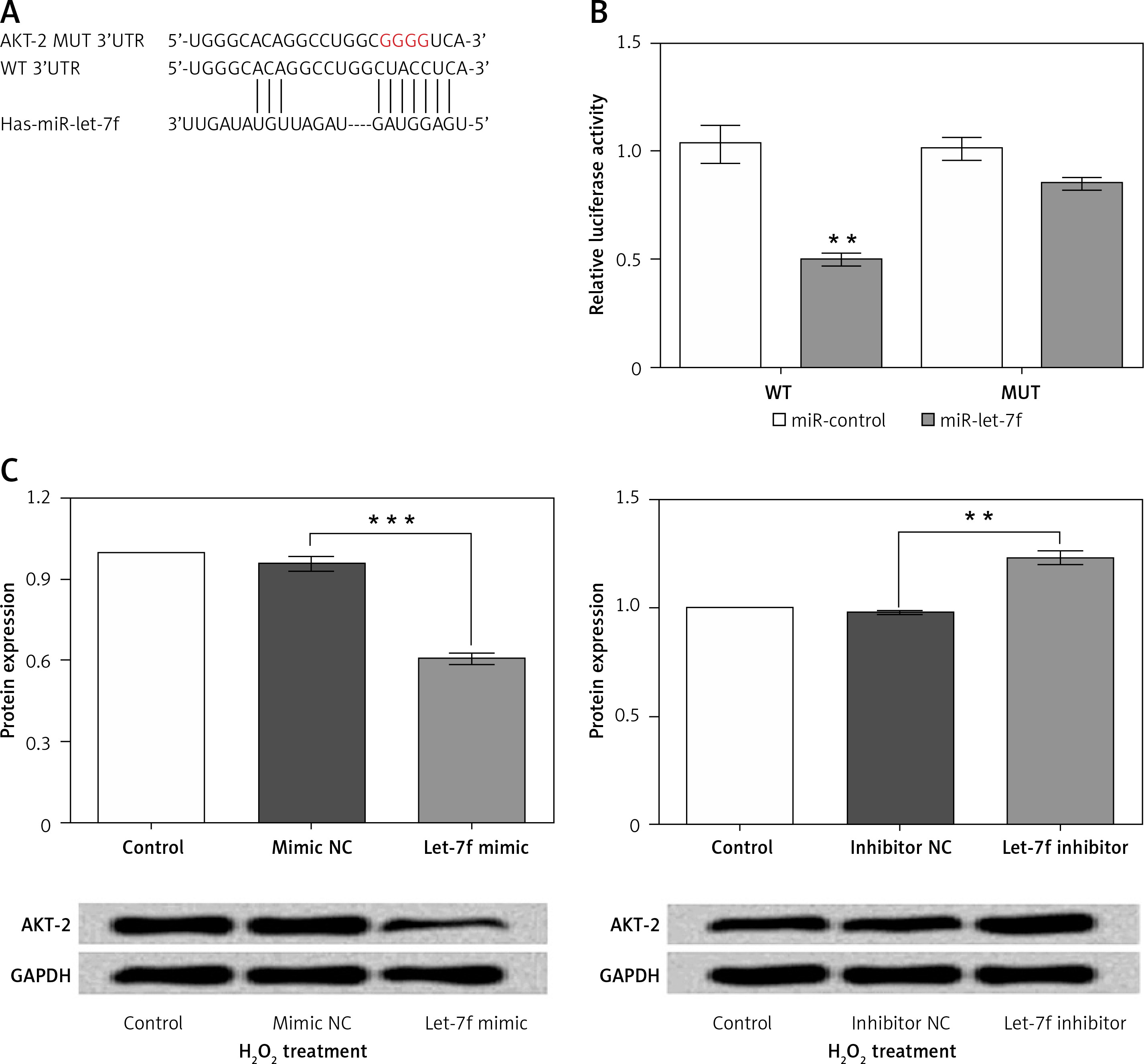
To determine the potential molecular mechanism of miR-let-7f in H2O2-induced oxidative damage, we performed a vast bioinformatic analysis to examine putative targets of the miR-let-7f. An Internet search was performed in various databases (microRNA.org and TargetScan) for its potential targets. As the bioinformatics analysis showed, the 3’-UTR of AKT-2 has consequential pairing with miR-let-7f, suggesting that AKT-2 was the potential regulatory target of let-7f (Figure 4 A). Then the luciferase reporter gene assay was performed to detect the relationship of miR-let-7f and AKT-2. The relative luciferase activity was significantly decreased in the SH-SY5Y cells co-transfected with miR-let-7f and the 3’-UTR of AKT-2 (p = 0.004, Figure 4 B). Our results suggested that AKT-2 is the direct target of miRNA-let-7f. Furthermore, we detected that expression of AKT-2 in H2O2-treated transfected SH-SY5Y cells using western blot assay. The results showed that the miR-let-7f up-regulation down-regulated the expression of AKT-2 (p = 0.0006), while the down-regulation of miR-let-7f enhanced the AKT-2 expression (p = 0.002, Figure 4 C). Overall, these results indicated that AKT-2 is the direct target of miR-let-7f, and was regulated negatively by miR-let-7f.
Figure 4
AKT-2 is a direct target of miR-let-7f and was negatively regulated by miR-let-7f. A – Alignment of miRlet-7f with AKT 3’-UTR sequences. B – Relative luciferase activity of reporters containing wild-type or mutated type with miR-let-7f target sites in H2O2-treated SHSY-5Y cells. C – miR-let-7f negatively regulated the AKT-2 expression
Data presented are the mean ± SEM (n = 3). **p < 0.01, ***p < 0.001.
Discussion
MicroRNAs are small, non-coding, regulatory RNAs that participate in posttranscriptional gene regulation in a sequence-specific manner [22]. Recently, several studies have indicated that the alterations of miRNA in the brain would contribute to neurodegenerative disease, including PD and AD [23, 24]. However, the molecular mechanism of miR-let-7f in AD pathogenesis has not yet been determined. In the present study, we identified candidate miRNAs that are differentially expressed in AD patients and healthy volunteers by miRNA microarray analysis. Among the 11 differentially expressed miRNAs, we focused on miR-let-7f because a previous study suggested that miR-let-7f had an anti-apoptotic role in an AD model [19]. Therefore, the present study focused the function of miR-let-7f in the H2O2-induced oxidative damage in vitro, and its potential molecular mechanism for the first time. The results demonstrated that miR-let-7f was overexpressed in H2O2-treated cells and its up-regulation protected against H2O2-induced oxidative damage in SH-SY5Y cells by negatively regulating AKT-2.
OS is a crucial factor in neurotoxicity associated with a variety of neurodegenerative diseases including AD, and H2O2-induced oxidative damage is a typical early feature of AD [5, 25]. Exposure to oxidative stress leads to the accumulation of intracellular reactive oxygen species (ROS), which are also associated with increased deposition of amyloid-β and formation of senile plaques, a hallmark of the AD brain [26, 27]. H2O2 is a major ROS in the ischemic brain and can be converted to hydroxyl radicals, which is specifically related to neurofibrillary pathology in AD [28, 29]. Thus, we induced oxidative damage in SH-SY5Y cells utilizing H2O2. As expected, when exposed to H2O2 in vitro SH-SY5Y cells showed significant apoptosis, decreased cell activity and induced cell morphological damage, accompanied by miR-let-7f up-regulation and increased apoptosis-related protein expression. Therefore, we studied the effects of miR-let-7f on H2O2-treated cells.
The miR-let-7f is a member of the let-7 family, which has been reported to be up-regulated in the AD patients and may be involved in regulating various neurodegenerative diseases [30]. In our study, we observed that miR-let-7f was overexpressed both in AD patients and H2O2-treated cells. These results indicated that miR-let-7f might participate in the pathogenesis of AD, which is consistent with the findings of a previous report [19]. Subsequently, we changed the expression of miR-let-7f to observe the effect of miR-let-7f on cell activity in H2O2-treated cells. Interestingly, up-regulating miR-let-7f in H2O2-treated SH-SY5Y cells increased cell viability, reduced cell apoptosis and alleviated the H2O2-induced morphological damage, indicating that overexpression of miR-let-7f partly alleviated the H2O2-induced oxidative damage in vitro. In Drosophila, miR-14 was reported for the first time to regulate cell proliferation and cell death [31]. During the past decade, multiple sources of evidences have also proved the involvement of miRNAs in regulating apoptosis and proliferation [32]. For example, this anti-apoptosis effect has also been reported in other let-7 family microRNAs, such as miR-let-7a, and miR-let-7b [33, 34]. Consistent with previous reports, we further identified the anti-apoptotic and pro-survival role of miR-let-7f in H2O2-induced oxidative damage.
Given the results of bioinformatic analysis, we examined the regulatory relationship between miR-let-7f and AKT-2. The results demonstrated that AKT-2 is the direct target of miR-let-7f, and was regulated by miR-let-7f negatively. AKT-2, as a key protein in activation of the PI3K/AKT pathway, serves an important role in regulating cell proliferation and apoptosis [35]. Additionally, the aberrant activation of apoptotic pathways has been reported in AD neurodegeneration, which indicates that it may be an early event in AD and contributes to the pathological processes of AD disease mechanisms [36, 37]. Therefore, we speculated that the overexpression of miR-let-7f in AD patients may activate the PI3K/AKT pathway by up-regulating AKT-2, and then inhibit the activation of apoptotic pathways. The other let-7 family miRNAs have also been demonstrated to play crucial roles in promoting cell survival by inhibiting protein kinase B or AKT [38, 39]. Overall, we presented a possible mechanism of miR-let-7f in AD pathogenesis whereby miR-let-7f protected against H2O2-induced oxidative damage by negatively regulating AKT-2.
Although some important findings were reported in the present study, there were several limitations in our study. MicroRNAs are extremely complicated and miR-let-7f alone cannot be responsible for the whole pathogenesis of AD. Thus, the detailed networks of AD pathogenesis remain to be further researched. Additionally, the present study was performed in cell lines in vitro, which may not fully reflect the biological process in real life. Therefore, it is necessary to verify the findings in an animal model.
In conclusion, we verified the overexpression of miR-let-7f in AD patients and proposed a possible mechanism of miR-let-7f in AD pathogenesis for the first time. The results demonstrated that the up-regulation of miR-let-7f in vitro alleviated the H2O2-induced oxidative damage by negatively regulating AKT-2. The aberrant expression of miR-let-7f may affect activation of the PI3K/AKT pathway. These findings provided a novel perspective in the role of miR-let-7f in pathogenesis of oxidative damage during AD and indicated that modified microRNA may be a promising approach for the therapy of neurodegenerative diseases.