Statins
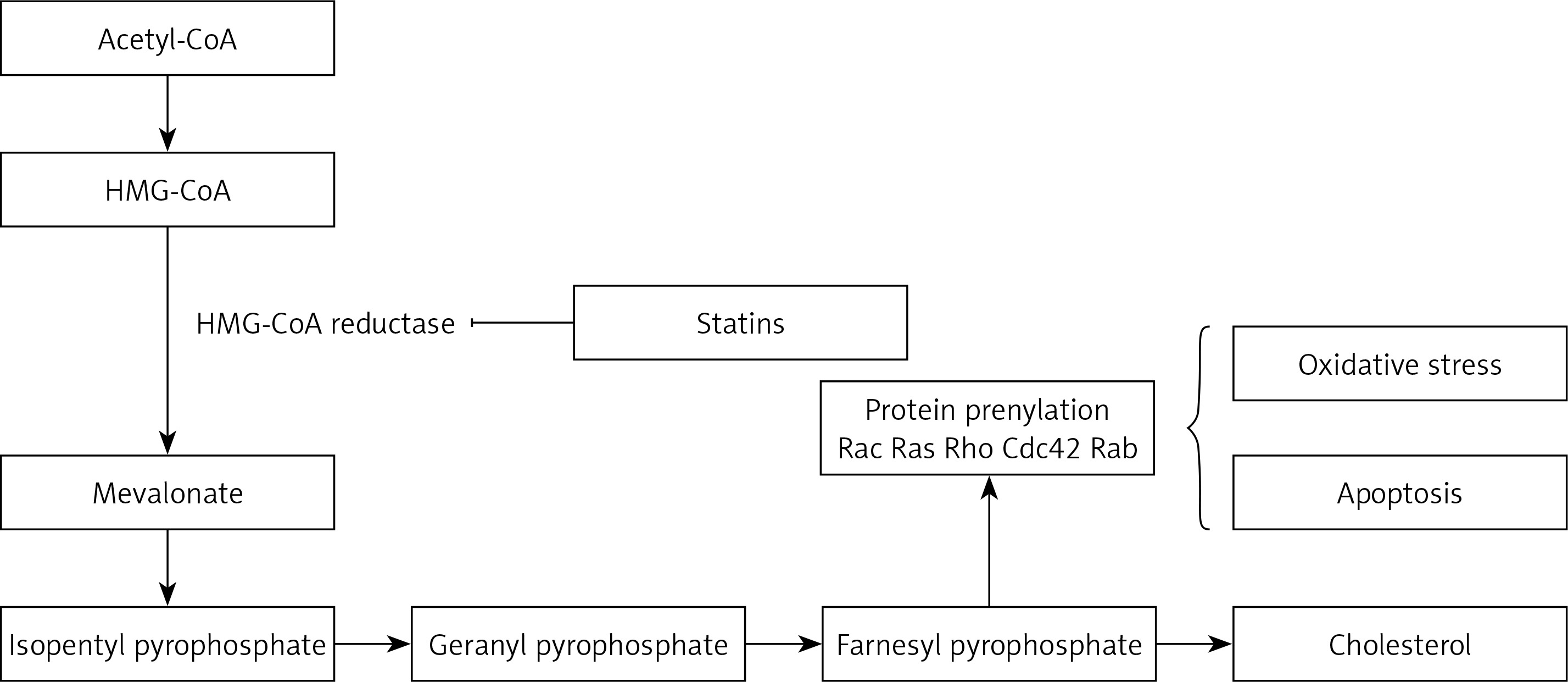
Statins are inhibitors of HMG-CoA reductase (HMGCR) and widely administered as lipid-modulatory agents in primary and secondary prevention of cardiovascular disease (CVD). HMGCR inhibition has a rate-limiting effect in the cholesterol synthesis pathway through conversion of 3-hydroxy-3-methyl glutaryl-CoA (HMG-CoA) to mevalonate. Statins are designed to match the enzymatic active site, resulting in structural changes, reduced enzyme activity and subsequent reduction in cholesterol synthesis [1].
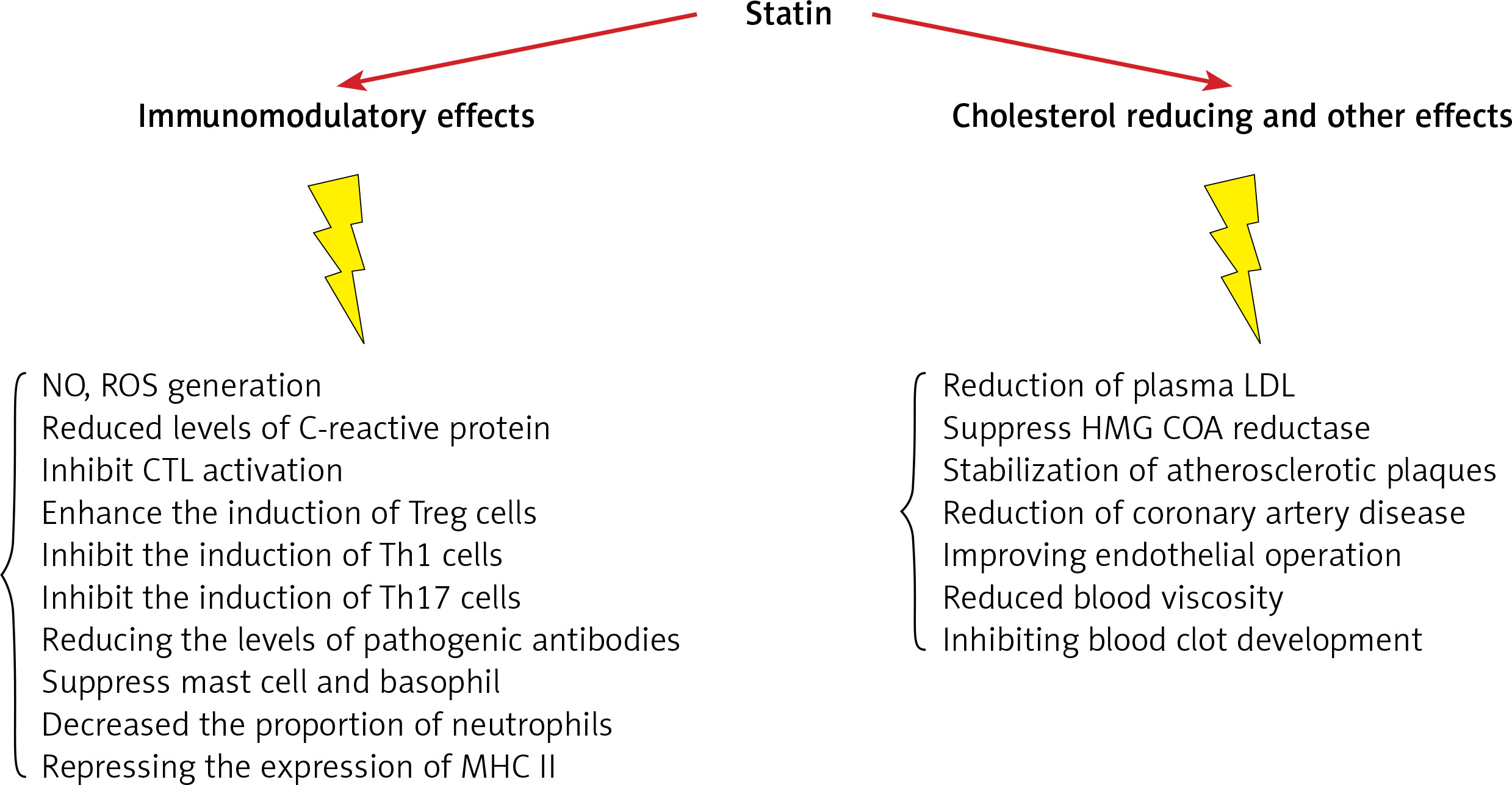
Statins decrease low-density lipoprotein (LDL) cholesterol levels more effectively than most of the other cholesterol-lowering drugs. Despite the introduction of newer cholesterol-lowering agents [2–5], statins are still the most widely used drugs for this purpose. They also reduce triglyceride and increase high-density lipoprotein (HDL) cholesterol levels [6–8]. Additional pharmacological effects include enhancing LDL uptake, reducing protein prenylation, improving endothelial function, modulating inflammation, preserving plaque stability and inhibiting thrombosis [9–22]. Overall, statins have a key role in cardiovascular treatment regimens reducing the risk of atherosclerotic diseases with different levels of effectiveness based on underlying risk factors and CVD history. Clinical practice protocols typically suggest in the first place lifestyle amendment, with potential benefits from adopting a cholesterol-reducing diet and increased physical activity. In those who struggle with lifestyle interventions or are unable to meet their lipid-lowering goals by these measures alone, statins may prove beneficial [23–26]. Their use is not without controversy and their advantageous therapeutic effects are sometimes overshadowed by reported negative effects. Prolonged use of statins can cause adverse effects such as myopathy, neurological effects, e.g. fatigue and impaired cognition, and enhanced diabetes risk [27–29].
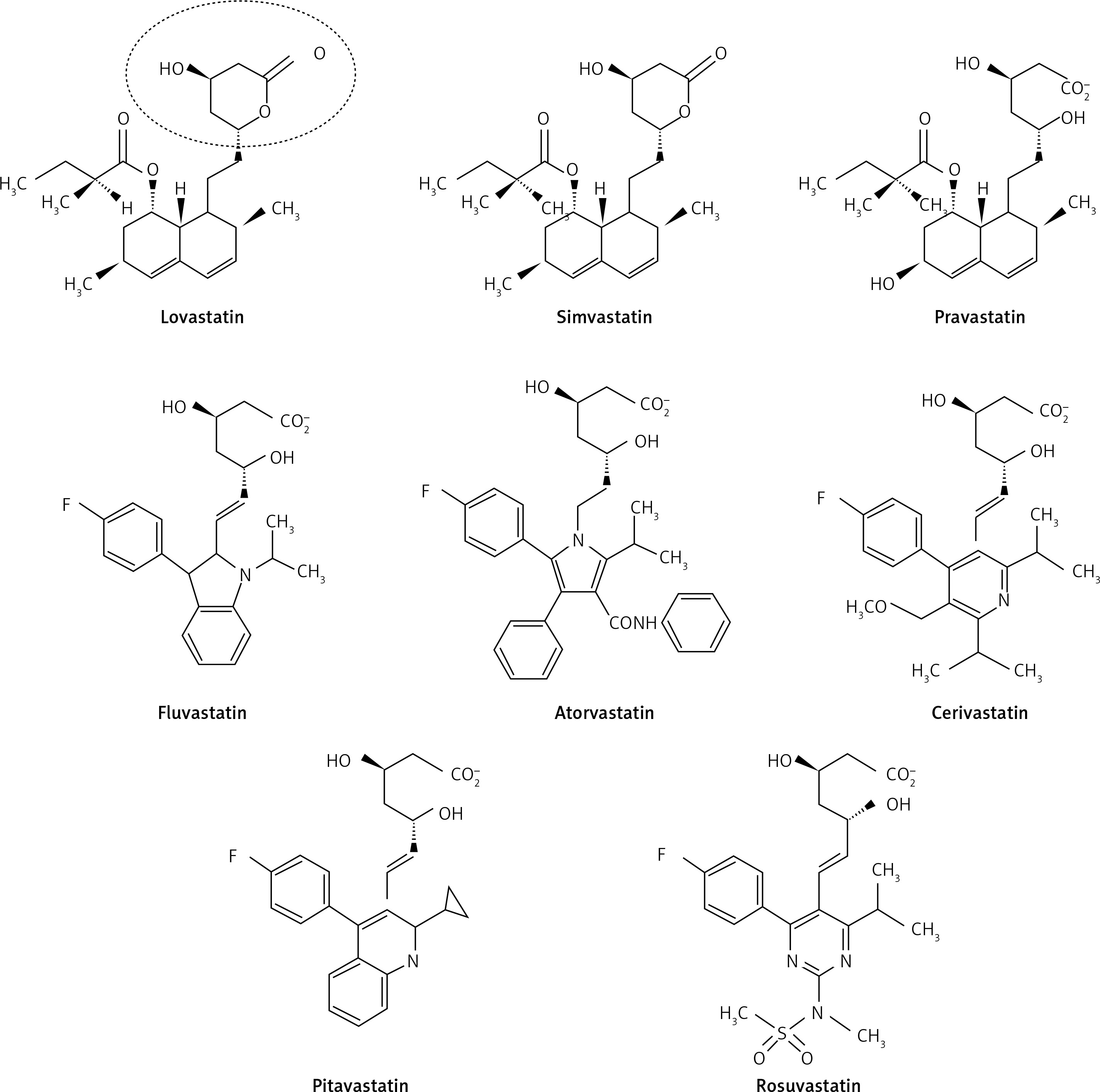
Statins are derived from two origins; 1. biological – extracted from Penicillium and Aspergillus fungal metabolites (examples include lovastatin, pravastatin and simvastatin); and 2. synthetic – including atorvastatin, cerivastatin, fluvastatin, pitavastatin and rosuvastatin [30, 31]. According to their lipophilicity they are divided into two groups: lipophilic statins including atorvastatin, simvastatin, lovastatin, fluvastatin, cerivastatin and pitavastatin which are susceptible to cytochrome P450; and hydrophilic ones including pravastatin and rosuvastatin, which are not susceptible to cytochrome P450 [32].
As with all medicinal classes, statins have similar mechanisms of action but vary in terms of chemical structure; therefore their pharmacokinetic properties and ability to modify lipid levels also vary. In terms of LDL-C lowering, rosuvastatin has proven most effective, whilst pravastatin and simvastatin are better for increasing HDL-C, and atorvastatin in reducing triglyceride levels [31, 32] (Figure 1).
Secondary to their important lipid-lowering effects, statins also display pleiotropic or non-lipid lowering effects. They have the potential to affect several immune responses including immune cell activation, migration, cytokine generation, immune metabolism, and survival. Statins also demonstrate immunomodulatory effects regulating innate and acquired immunity through modulating interactions of T-cells and antigen presenting cells (APCs), thus altering inflammatory cytokine levels. They could affect the volume of red blood cells (RBC) and platelets (PLT) as well. In the following section their effects on blood cells will be discussed.
Blood cell differentiation
The changing of cells from one type to another is called cellular differentiation [33, 34]. The cell usually converts to a more specific kind (Figure 2). Differentiation happens many times throughout the evolution of a multicellular organism while it switches from a simple zygote to a complicated system of tissues and cell groups (Figure 2). Stem cells are divided during tissue repair and cellular circulation, and produce completely differentiated daughter cells, with differentiation maintained in adult cells. There is some differentiation when an antigen is located on the cell surface. Size, structure, membrane potential, metabolic function and signal reflection of a cell are radically modified by differentiation. These modifications are attributable to extremely controlled changes in gene expression (understood by epigenetics). Except for some cases, cell differentiation never results in a change in the DNA sequence. Therefore, even with a similar genome, cells have potentially numerous physical differences [35].
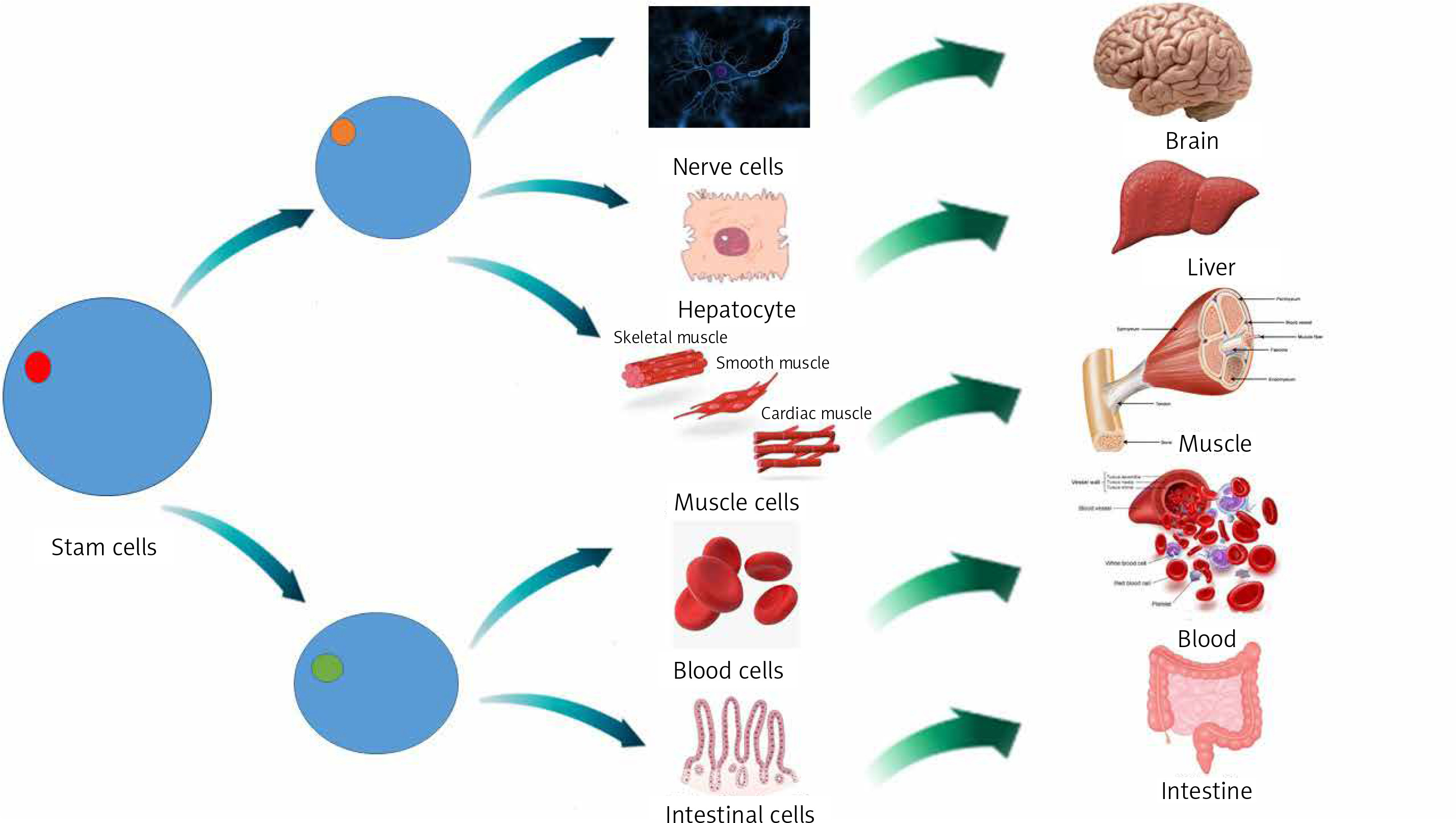
Mesenchymal stem cells (MSCs) are tissue-resident multipotent stem cells that have demonstrated the ability to differentiate into various cell lines, including mesodermal adipogenic, chondrogenic, osteogenic hepatogenic, ectodermal neurogenic lines and, also into blood cells [36].
MSCs are disseminated throughout all tissues and aid tissue repair and homeostasis. At first MSCs are commonly discovered in a dormant phase and it is proposed that they re-enter the cell cycle when stimulated internally or externally and progress towards proliferation and differentiation [37]. This primary contact of MSCs with other cell types is essential for ordinary tissue homeostasis. For instance, it was revealed that a mixture of endothelial cells with MSCs is engaged in the development of vascular networks [38]. Moreover tissue-resident stem cells can differentiate into macrophages (MQ), as demonstrated by gene expression, cell surface marker attributes, cytokine manufacture or even functional behavior [39].
Effects of statins on cells which play roles in immunity
T cells
As mentioned above, statins may be immunomodulatory and can affect T cells. Two different pathways have been suggested in relation to this action; an HMGCR-independent pathway, including allosteric hindrance of lymphocyte function-associated antigen 1 (LFA-1), which represses activation, proliferation, and cellular migration of T cells [40] and HMGCR-dependent mechanisms, which rely on their ability to prevent prenylation of small GTPases [41, 42]. Moreover, new records have presented data that some statins change T helper cell differentiation through developing T helper 2 polarization and repressing T helper 1 polarization both in vitro and in vivo [43, 44]. Work by Arora et al. showed that simvastatin develops T helper 2 cell polarization through up-regulation of the chitinase family member Ym1 in dendritic cells (DCs) [45]. Moreover, statins prevent T cells from differentiating by interfering with the function, differentiation and maturity of APCs. Statins impede APC development through down-regulation of CCR7, CD40, CD83, and CD86 on cytokine-excited DCs [46].
CD8+ T-cells
Deficient T-cell homing is associated with an increased inability of T cells to interact with peptide antigens offered by APC, a mechanism that happens in peripheral lymphoid organs. However, statins have been reported to prevent the activation and proliferation of T cells interacting with agonistic anti-CD3 monoclonal antibody (mAb) or Staphylococcus aureus enterotoxin (SEB), an impact attributed to the deficiency of pathways regulated by small Ras-like GTPases in T-cell receptor (TCR) signaling for the former stimulus through lymphocyte function-associated antigen 1 (LFA-1) blockage [47, 48]. Bu et al. discovered that simvastatin or pitavastatin significantly hinders proliferative responses of immature murine cytotoxic T lymphocytes (CTL) for TCR stimulation due to Kruppel-like factor 2 (KLF2). They furthermore found that statins negatively affect the effectors of CTL proliferation and IFN-γ expression. These findings indicate that statins can affect both the priming and the effector stages of T-cell responses. Simvastatin and pitavastatin also hindered the modest proliferative response of T cells triggered by α-CD3, and more vigorous proliferative response of human T cells to αCD3 and anti-CD28. Human T cell IFN-γ up-regulation, resulting from PHA plus IL-2 stimulation, has also been drastically decreased by simvastatin. Decreased IFN-γ expressed by human T-cells induced by PHA and IL-2 was also demonstrated [49].
As outlined above, statins disrupt processing and presentation of antigens by MHC class II in human DCs and B lymphocytes via reduced antigen uptake by both receptor-mediated endocytosis and macropinocytosis [50]. Activation of Rho and Rab GTPases leads to the primary phase of extracellular antigen cross-presentation in which antigens are absorbed through phagocytosis or receptor-mediated endocytosis [51, 52]. Moreover, hydrophilic proteins need localization into specific organelles defined by the expression of Rab5 and EEA1 to be cross-presented [53]. It is proposed that statins disrupt this process, as in simvastatin-treated APCs, cross-presentation was dramatically repressed. This repression was inverted by adding mevalonate, demonstrating that simvastatin’s repressing influence on cross-presentation depends on its HMGCR inhibiting ability [54]. Mannose receptor endocytosis of particulate antigens is crucial for cross-presentation [53]. To determine simvastatin’s influence on mannose receptor-dependent endocytosis, absorption of fluorescein isothiocyanate (FITC) labeled dextrans, which are internalized through both fluid-phase and mannose-dependent endocytosis, was evaluated [55, 56]. Simvastatin treatment led to a considerable decrease in splenocyte absorption of dextran-FITC, an impact that was modified by mevalonate. While simvastatin can prevent extra levels in antigen cross-presentation through APC, poor absorption of soluble antigen seems to be key in cross-presentation malfunction.
Statins and Treg cells
There is increasing evidence aiding understanding of the mechanisms behind the immunomodulatory effects of statins from in vivo and in vitro investigations. This includes proposed up-regulation of endothelial nitric oxide synthase (NOS3), decreased stimulation of endothelial cells, development of anti-oxidative responses, decreased migration of inflammatory cells, decreased antigen presentation because of suppressed MHC II expression in endothelial cells [57] resulting from promoter IV of class II trans-activator (CIITA) [58], down-regulation of pro-inflammatory cytokines, decreased PLT accumulation, development of 15-epi-lipoxin (15-epi-LXB4), which operates through preventing the signaling pathways engaged in the stimulation of the inflammatory response induced by lipid agents [59, 60] and, finally, enhanced migration, differentiation, and prohibitory functions of Treg cells [59, 61]. It is well affirmed from recent experimental findings that interleukin 6 (IL-6) signaling via STAT-3 contributes to repression of foxp3 transcription, so the probable repression of IL-6 signaling by statins may indirectly support foxp3 expression, leading to an improved proportion of Treg cells [62].
Statins and Th1 cells
It is proposed that statins affect Tregs’ operation through suppressing T helper 1 immune reactions. In accordance with this, statins can regulate various stages of T helper1 induction. Another mechanism includes modulating CD40 expression in addition to recognition on developed B cells and some epithelial cells. It stimulates IL-12 expression, facilitating differentiation and clonal expansion of TH cells toward T helper1 cells [63]. Atorvastatin downregulates CD40 and IFN-γ induced generation of TNF-α on human endothelial cells and monocytes; this effect is altered by administration of exogenous mevalonic acid. In addition to HMGCR impairment, statins also alter differentiation of white blood cells in T helper1 and T helper17 phenotypes, resulting in development of regulatory T cells which express the Foxp3 transcription factor in vitro and in vivo [64]. Also, utilizing a geranylgeranyl transferase inhibitor (GGTI-289) and farnesyltransferase inhibitor (FTI-227), GGTI-289 but not FTI-227 imitates the impact of simvastatin, recommending that hindrance of geranylgeranylation protein dictates the decreased differentiation of T helper17 cells and the expanded differentiation of Treg cells [65].
Statins and Th17 cells
Research on murine models (BALB/c mice) by Li et al. indicated that simvastatin suppresses T helper17 cell differentiation and improves regulatory T cell differentiation in spleen cells grown under varying polarizing circumstances. The researchers also observed that simvastatin treatment in mice reduced infiltration of inflammatory cells toward the peripheral nervous system (PNS) and reduced the rate of IFNγ and IL17 constructing lymphocytes [66]. Moreover, atorvastatin treatment led to downregulation of co-stimulatory molecules for T-cell stimulation, for example, CD80, and enhanced the number of regulatory T cells in mononuclear cell lymph nodes [66]. Accordingly, this information suggests that atorvastatin may work as a suppressor of inflammatory T helper-1 and -17 reactions, downregulating co-stimulatory molecules and concentration of Tregs.
Statin and B cells
Atorvastatin-stimulated naive DC to stem cell conversion in vitro was demonstrated by Li et al. Treatment of these tolerogenic DCs improves experimental autoimmune myasthenia (EAMG) via repressing expansion of lymphocyte cells, development of Treg cells, transformation of T helper1 and T helper17 to T helper2 cells and reducing pathogenic antibody levels [67]. However, the defined systems by which tolerogenic DC treatment decreases antibodies were not completely comprehended. Li et al. discovered that immature bone marrow-derived dendritic cells (BMDCs) were effectively stimulated by atorvastatin under laboratory conditions, which in turn suppressed CD80, CD86, and MHC II molecules [68]. Atorvastatin-modified BMDCs (AT-BMDCs) repressed the number, and the relative affinity, of antiR97-116 IgG antibodies’ further germinal center (GC) function. In addition, T-follicular helper (Tfh) cells and IL21 were reduced by AT-BMDC treatment. Furthermore, the level of CD19+ B cells was somewhat affected, while regulatory B cells were enhanced via AT-BMDCs. One may conclude that AT-BMDCs might improve myasthenia gravis (MG) by controlling humoral immune function and could present a possible approach for MG treatment.
Statins and mast cell and basophil IgE
Kolawole et al. reported that a variety of statins hinder IgE-incited cytokine generation of mast cells and basophils with differing levels of effectiveness [69]. In addition, to prevent fluvastatin repressing vascular reactions to histamine, mice were pre-treated with fluvastatin and by infusing histamine the activity of mast cells was inhibited, which created conditions in which fluvastatin had no effect. This confirms the hypothesis that mast cells are inhibited in vivo by fluvastatin, and supports the use of statins for treatment of mast cell-related disorders. Kolawole et al. also studied the effects of fluvastatin on both C57 black 6 (B6) and 129/SvImJ (also called 129) basophils [69]. They demonstrated that culture for 24 h with fluvastatin before IgE+antigen-mediated activation decreased IL-4, IL-6, IL-13, TNF, and monocyte chemoattractant protein-1 (MCP-1) construction, and prevented degranulation among B6 basophils, but had no major consequence for SvImJ basophils [69].
Statins and neutrophils
The most plentiful type of blood granulocytes is neutrophils, which play an important role in protecting the body against pathogens [70]. Statins interfere with these as outlined earlier, by repression of HMGCR, inhibiting the transformation of HMG-CoA to mevalonate with the downstream restraint of cholesterol construction [71]. Also, metabolites that are constructed during the formation of cholesterol are utilized in other cell-signaling pathways. For instance, some products of the cholesterol synthesis pathway, such as geranylgeranyl pyrophosphate (GGP) and farnesyl pyrophosphate (FPP), are included in the prenylation and subsequent activation of the small GTPase group of signaling molecules including RhoA with its effector Rho kinase [72]. Maher observed that pravastatin and simvastatin decreased the proportion of neutrophils that moved through collagen-coated filters [73].
Statin treatment has been revealed to promote overall longevity, decrease the prevalence of graft vasculopathy and decrease the intensity of cardiac allograft rejection incidents [74, 75]. Furthermore, statin use after lung transplantation is correlated with less acute rejection episodes, reduced bronchoalveolar lavage (BAL) neutrophil numbers and enhanced 6-year survival [76]. Chello et al. also found that atorvastatin greatly decreased expression of neutrophil CD11b and neutrophil-endothelial adhesion in patients before cardiopulmonary bypass (CPB) [77]. Simvastatin has additionally been discovered to weaken fMLP-stimulated IL-8 secretions from neutrophils, in addition to considerably decreasing ROS production [78]. Cerivastatin and pravastatin also appear to restrain neutrophil transendothelial movement [79].
Statins and eosinophils
Statins can influence chemokines, e.g. IL-8, IL-12, IL-17, IL-6, and MCP, and adhesion molecules such as CCR7, CD40, LFA-1, CD83, and CD86, thus interfering with the migration of inflammatory cells from the blood into airways. Because eosinophils and MQs both express LFA-1 molecules, this suggests a possible goal for treatments targeting airway inflammation. Medications besides statins targeted at down-regulation of LFA-1 expression have been useful in reducing airway eosinophil count in murine models of allergic asthma [80], and have decreased sputum eosinophil count following allergen challenge in asthmatics [81]. Statins have an ability to prevent LFA-1/ICAM-1 correlation, like those observed in AIDS [82]. There is potential for an equivalent effect in asthma where the etiology is correlated with eosinophil aggregation. The perception that statins enhance eosinophil apoptosis in individuals [83] suggests an additional curative function. This is likely attributable to the quick decrease of cellular CD40 expression following statin treatment, which completely suppresses eosinophil survival [84].
Statins and macrophages
Statins are typically prescribed long term; it is therefore necessary to realize the impact of persistent statin administration, and how they influence biological features of stem cells. Izadpanah et al. [85] administered MSCs with atorvastatin or pravastatin at clinically appropriate levels and evaluated MSC generation, differentiation possibility, and gene expression profile. Atorvastatin and pravastatin decreased the overall development rate of stem cells. Primarily, statins decreased the potential of stem cells to differentiate into MQs as they showed no direct impact on MQ activity. The conclusions were that MSCs’ insufficient ability to differentiate into MQs may lead to reduced MQ frequency inside the arterial plaque, decreased inflammation, and afterward increased plaque stability. This reveals the non-lipid-related decrease in CVD. In terms of adverse effects, statins reduced stem cells’ osteogenic and chondrogenic differentiation potential and enhanced cell senescence and apoptosis, as shown through p16 (CDKN2A), p53 (TP53) and caspase 3, 8 and 9 upregulation. Statins also downregulate DNA repairing genes, containing X-ray repair cross-complementing protein 4 (XRCC4X), XRCC6, and Apex1. As the impact on MQ differentiation highlights the advantageous side of statins, their influence on other biologic features of stem cells presents an original description of their unfavorable clinical consequences.
Statins and human monocyte-derived DCs
Kwak et al. [86] stated that statins repress MHC expression on human MQs and endothelial cells stimulated by IFN-γ, resulting in a decreased capability of these cells to stimulate T cells. The priming of mice naive T cells with spleen APC in the presence of statins resulted in prevention of Th1 cell differentiation [43]. Moreover, atorvastatin treatment reduced the expression of HLA-DR and CD38 in healthy people [87]. Statins primarily repress the expression of MHC-II antigens on human MQs, endothelial cells, and smooth muscle cells (SMC) excited by IFN-γ [86, 88]. This result was applied by different kinds of statins, lipophilic and hydrophilic, at nanomolar to micromolar levels, but it was restricted to APC cells needing co-stimulation via IFN-γ. Professional APC cells, such as B lymphocytes, and DCs which fundamentally express MHC class II molecules, were not affected.
DCs are disseminated everywhere and demonstrate the specific strength to trigger a basic immune response through naive T cell stimulation [89]. In order to have the capability to activate T cells, DCs are matured by increasing surface MHC class II and co-stimulatory molecule expression. DCs have been observed in atherosclerotic plaques, confirming their involvement in arteriosclerotic vascular disease [90, 91]. Moreover, some atherogenic agents such as LDL, cholesterol [92], LPC [93], nicotine [94], and angiotensin II [95] facilitate DC maturation. Research has also shown that development of DCs in atherosclerotic plaques, specifically in colocalization with T lymphocytes, relates to plaque developments and ischemic syndromes, outlining DCs’ role in plaque instability as a result of triggering inflammation [96]. In the study of Yilmaz et al. some mature DCs and a higher constant plaque morphology were identified in atherosclerotic plaques of cases treated with a prevalent statin (simvastatin or atorvastatin (0.1–1 µM)). For this reason, we presumed that DC movement into atheroma could be decreased via statins [97]. Researchers have revealed that statin treatment reduces DC adhesion and migration across the endothelial monolayer [98]. It has also been demonstrated that abnormal levels of cholesterol and other lipids induce a decline in DC migration from atherosclerotic plaques into environmental lymph nodes, resulting in their local enhancement [99].
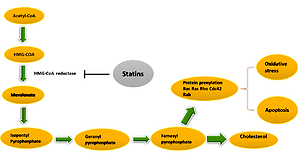
Statin effects on the endothelium might be related both to their hypolipidemic effects and or direct effects on endothelial cells. For instance, intracellular signals of Rho-kinases, Rac, and Cdc42, which precipitate prothrombotic and anti-inflammatory activity of endothelial cells, are suppressed by statins (Figure 3) [59]. resulting in reduced oxidative stress and inflammation. Furthermore, statins can remarkably enhance cellular communication network factor 3 (CCN3) expression, prevent expression of pro-inflammatory VCAM-1 cell adhesion molecule, and inhibit monocyte adherence to endothelial cells through the KLF-2 transcription factor pathway, lowering endothelial inflammation [65].
Statins and platelets and RBCs
In the study of Xian-Yu et al., patients were administered atorvastatin or simvastatin. Lipids and hematological variables were assessed as well as risk factors for CVD at baseline and 4-week treatment points. The data showed that lipids were modified considerably following 4 weeks’ treatment, mean platelet volume (MPV) dropped dramatically for hematological variables, and the decrease was similar between the two statins. There was no association between MPV and lipid changes [100]. RBC deformation is associated with cell release of ATP [101]. It has been demonstrated that membrane cholesterol changes the features of the cell membrane, e.g. fluidity and bending rigidity [102], and in some CVDs, enhanced membrane cholesterol is found. Therefore, one of the mechanisms contributing to the antithrombotic effects of statins is their induced reduction in the structure of the erythrocyte membrane lipid that enhances RBC deformation and then reduces accumulation of RBC. Xian-Yu reported that MCV discrete degree (SD) became higher following 1 month of statin treatment, 93.1 ±1.42 vs. 93.4 ±2.72 for simvastatin, 93.1 ±1.08 vs. 94.4 ±3.51 for atorvastatin, and substantial variations were observed between before and after atorvastatin treatment. Statins may influence the volume of RBC; however, the extent of this varies between statins. Clapp et al. reported that simvastatin administration to rats or incubation of healthy human RBCs with geranylgeranyl transferase inhibitor-2133 (GGTI-2133) enhanced RBC deformability but diminished low O2 tension-induced ATP release. This suggests that unlike the Rho kinase inhibitor Y-27632, that enhances RBC deformation and raises the release of low O2 tension-induced ATP, simvastatin blocks low O2 tension-induced ATP release through uncoupling RBC deformation and low O2 tension-induced release of ATP. Accordingly, in vivo, simvastatin administration can interfere with the erythrocyte’s strength to cooperate in optimizing blood perfusion distribution to meet O2 tissue needs in the skeletal muscle [103]. In addition, treatment with rosuvastatin dramatically enhanced phosphorylation of NOS, NO-forming dependent on NOS, and deformability of erythrocytes. Rosuvastatin’s stimulating impact on RBC-NOS function was reversed by the NOS inhibitor NG-monomethyl-L-arginine. This NO-dependent impact of rosuvastatin may have a significant effect on the microcirculation and can offer new potential for statin therapy application in the clinic [104].
Conclusions and future perspectives
Research to date proves that statins have important anti-inflammatory and immune-modulatory features that may be relevant to their effectiveness in CVD prevention and remedy (Figure 4, Table I) [105–119]. Statins have several effects on the differentiation and function of immune cells. They increase the amount of Treg cells and inhibit Th17, Th1, Th2 and CTL cells. They also have a large effect on various cytokines, such as IL-17, IL-6, IL-4, IL-23, IL-10, IL-21. It has also been proven that statins lead to the suppression of mast cells and basophils and reduce antibody levels. Physicians should be aware that combining statins with other immunomodulatory drugs requires caution as dual therapy may precipitate reactivation of latent infectious diseases and drive tumor progression. Given the proposed wider application of these medicines and their potentially important advantages in treatment of inflammatory and autoimmune disorders, more studies are needed with special focus on the molecular targets of statins included in regulating the immune response. This will allow further conclusions to be met and build an evidence base for wider application of statins in the clinic.
Table I
Evidence on immunomodulatory effect of statins in humans and mice
| Statin | Dose | Experimental model | Possible mechanism | Ref. |
|---|---|---|---|---|
| Simvastatin | In vitro: 2 µM | Mice, Treg/Th17 differentiation | ↑ Differentiation of CD4 into Treg ↓ Differentiation of Th17 ↑ SOCS3 | [105] |
| Simvastatin | In vitro: 5 µM | Human | ↑ IL-4, IL-5, IL-13 ↓ INF-γ ↓ T-bet expression ↑ GATA 3 and B220 expression | [106] |
| Simvastatin | In vitro: 10 nM | Human, PBMCs from MS patients and healthy controls | ↓ IL-6 and IL-23 ↑ INF-γ, IL-4, and Il-27 ↓ RORC and IL17 gene expression ↑ SOCS3 | [107] |
| Simvastatin | 10, 50 mg/kg | Mice, tumor growth of 3LL cell line | ↑ Fox-p3, IDO, IL-10, and TGF-β | [108] |
| Simvastatin | In vitro: 10 mM | Human, PBMC from SCA patients | ↑ Treg cells’ number ↑ Treg cells’ suppressive activity | [109] |
| Simvastatin | In vitro: 10 mM | Human , monocytes | ↑ SOCS 3 and 7 ↑ INF-γ, IL-4 and IL-27 ↓ JAK-STAT expression ↓ IL-6, IL-23 and IL-17 | Reference |
| Simvastatin | In vitro: 10 µM | Human, mDC from asthmatic patients | ↓ Th17 cells, ↓ IL-6, ↓ IL-23 ↑ Treg cells ↑ IDO and ↑ IL-10 | [110] |
| Simvastatin | In vitro: 0.5–2 µM | Mice, Treg differentiation | ↑ Differentiation of Treg ↑ Foxp3 expression ↓ SMAD6 and SMAD7 ↓ Methylation of foxp3 promoter | [111] |
| Fluvastatin, lovastatin, and simvastatin, | In vitro: all at 10 µM | Mice, T cell | ↑ Kruppel-like factor 2 (KLF2) expression ↓ INF-γ ↓ Differentiation of T-cells | [49] |
| Rosuvastatin | 5 mg/kg i.v. | Mice, Ischemia-reperfusion injury | ↑ Foxp3 expression | [112] |
| Fluvastatin | 0.1, 1.0, or 10 µM | Human, PBMCs from 7 patients with asthma | ↓ IL-5, IFN-γ, CCL17, and CXCL10 Prevention of Th1 and Th2 cell migration | [113] |
| Lovastatin | 10 mg/kg i.p. | Mice, DTH (C. albicans) | ↑ Migration of Tregs to the foot-pad ↑ CCl1 and IL-10 | [114] |
| Lovastatin | 6.25, 12.5, 25 µM | Human | ↑ Bak expression (apoptosis regulators) ↓ Bcl-XL expression activation of caspase-3/8/9 ↓ Collagen and platelet aggregation | [115] |
| Lovastatin | 0.5–25 mM | Human, PBMC from patients with CIU | ↓ T-cell proliferation ↓ IL-10 and IL-17 ↔ Neither Socs3 nor RORc | [116] |
| Atorvastatin | 10 mg/kg | Mice, experimental autoimmune glomerulonephritis | ↑ IL-10, ↓ IL-17, IFN-γ ↓ TNF-γ IL-21 Treg | [117] |
| Atorvastatin | 20 mg/kg | Human, twenty-seven healthy volunteers | ↓ HLA-DR and CD38 expression ↑ Superantigen-mediated T cell activation | |
| Atorvastatin | In vitro: 10 mM | Human endothelial cell culture | ↓ Expression of CD40 | [64] |
| Atorvastatin | 10 mg/kg/day | Mice with EAE, control mice | ↑ Th2 cytokine secretion ↓ Th1 cytokine secretion | [118] |
| Atorvastatin | In vitro: 10 mM | Mice, experimental autoimmune myasthenia gravis | ↑ Foxp3 expression, ↑ Treg number ↓ Lymphocyte proliferation ↓ Th1/Th17, ↑ Th2 | [67] |
| Atorvastatin | In vitro: 5–10 mM | Human, PBMC culture | ↑ Treg cells’ number ↑ Suppressive function of Tregs | [119] |