Introduction
Cerebral ischaemic stroke has long been recognised as a prevalent and serious neurological disease associated with high mortality and morbidity [1]. The pathophysiology of ischaemic stroke is complicated and multifactorial [2–4]. There is an urgent need to determine the mechanisms behind stroke pathophysiology. Accumulating evidence has suggested that neuronal apoptosis plays an important role in cell loss following acute ischaemic damage through the activation of various signalling pathways [5–7]. However, the precise mechanism that regulates the activation of apoptotic pathway in the injured brain is yet to be fully elucidated.
FoxO3a, member of the forkhead box O (FoxO) transcription factor family, is a downstream transcriptional factor of the PI3K/AKT pathway [8–12]. Once it is phosphorylated by AKT, FoxO3a rapidly translocates from the nucleus to the cytoplasm, where phosphorylated FoxO3a is ubiquitylated and degraded [13–17]. Moreover, FoxO3a can be phosphorylated by IKK and ERK as well as in response to growth factor and insulin stimulation [15, 18]. Taken together, negative regulation of FoxO3a by phosphokinases is a key mechanism of promoting FoxO3a functions, but downstream molecular events remain elusive.
FoxO3a has multiple functions, most of which are related to ischaemia/hypoxia stress and tumour regulation. FoxO3a was first discovered due to its role in tumour differentiation and proliferation [16, 19–22]. However, the role of FoxO3a in ischaemia and hypoxia is controversial. Experimental cardiac research indicates that FoxO3a causes cardiomyocyte death [23, 24]. Another study showed that FoxO3a has protective effects in cardiomyocyte ischaemia-reperfusion (I-R) [25], and similar reports have been made in the kidney [26], liver [27], and lung [28]. At present, there have been few studies on the role of FoxO3a in cerebral ischaemia/hypoxia, especially in I-R. The total amount of FoxO3a is unchanged when cerebral ischaemia and hypoxia occur, but the level of dephosphorylated FoxO3a (d-FoxO3a) increases, leading to the entry of FoxO3a into the nucleus, which further results in an increase in apoptosis-related proteins and thereby promotes apoptosis [29–31]. It has also been suggested that I-R changes FoxO3a levels in the hippocampal CA1 region [32]. Protein expression of phosphorylated FoxO3a (p-FoxO3a) decreased during cerebral ischaemia and hypoxia but increased 4 h after reperfusion [33]. Considering the complex biological behaviour of FoxO3a in ischaemia/hypoxia-related cerebral injury, we aim to investigate its underlying mechanisms.
CircFoxO3 is a newly identified circRNA generated from gene FOXO3, which locates on chromosome 6q21. CircFoxO3 was reported to be downregulated in breast cancer [34] and non-small cell lung cancer [35] but upregulated in glioblastoma [36]. Functionally, circFoxO3 acts as a powerful tumour regulator through sponging certain miRNAs targeting the parental transcript, FoxO3 [37]. Considering the multiple functions of protein FoxO3a in tumour regulation, circFoxO3 may act as suppressor or activator depending on certain tumour types. William et al. reported that circFoxO3 was upregulated in heart samples from aged patients [38] and that it promoted cardiac senescence. However, the role of circFoxO3 in ischaemic stroke, which has not been reported, is of interest.
In the present study, we investigated whether the expression levels of FoxO3a and circFoxO3 changed during I/R. We further explored whether FoxO3a protected hypoxic neuronal cells against apoptosis, as well as its synergistic effect with circFoxO3.
Material and methods
Animals and groups
Male Sprague-Dawley (SD) rats (purchased from the Experimental Animal Centre of Renji Hospital, Shanghai Jiaotong University School of Medicine) weighing 250–300 g were housed in a climate-controlled room, with five rats per cage, and with food and water provided ad libitum on a 12 : 12 h light : dark cycle. The animal study protocol was approved by the Ethics Committee for Animal Experiment and conducted according to the Guidelines for Animal Experimentation of Renji Hospital, Shanghai Jiaotong University School of Medicine (20170223-001). Non-retrospective ethical approval for the animal experiments was obtained for the study. A total of 18 rats were randomly assigned to the following groups: sham group (operated on but not subjected to occlusion), MCAO group (operated on and subjected to occlusion for 90 min), and MCAO/reperfusion group (operated on and subjected to occlusion for 90 mins before being reperfused for 30 min).
MCAO/reperfusion model construction
Briefly, SD rats were intraperitoneally anaesthetised with 50 mg/kg pentobarbital sodium (VWR International, Ltd., Poole, England) and then attached to the operating table. The external carotid artery (ECA) and its branches were isolated and ligated, and a 3-0 monofilament nylon suture (Beijing Shadong Industrial Corp., China) with a heat-treated, rounded tip was introduced into the right internal carotid artery (ICA) via the ECA until slight resistance was achieved. The incisions were then sutured, and the rats were left alone for 90 min. After that, the rats were anaesthetised again as described above before the suture was withdrawn to facilitate reperfusion. The rats were finally euthanised with excess pentobarbital sodium, and whole brain tissues were removed. The extracted brain tissues were program prepared into paraffin sections before assessment. The area of the infarct volume was analysed using Adobe Photoshop CS4 Extended Version 11.0.1 (Adobe Systems Inc., San Jose, CA, USA). The infarct volume of each slice was calculated using the following equation: infarct volume = infarct area of the slice × thickness of the slice (2 mm). The total infarct volume was calculated by adding all the infarct volumes of each slice.
Cell culture and treatment with CoCl2 to mimic hypoxia
The rat neuroblastoma B35 cell line (ATCC, Manassas, VA) has been established as a suitable model to study hypoxia-injury (HI)-induced neuronal injury [35, 39]. The cells were maintained at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Carlsbad, CA, USA) containing 10% heat-inactivated foetal bovine serum (Gibco) in a humidified atmosphere with 5% CO2. CoCl2 was diluted in DMEM and sterilised through a 0.2 micron filter prior to use. According to previous studies, CoCl2 was further diluted in DMEM to a final concentration of 250 µmol for use in subsequent experiments [35, 39]. The cultures were divided into control (0 h) or CoCl2-treated (treated for 24, 48, or 72 h) groups.
Reverse transcription-quantitative PCR
Total RNA was extracted from the cells or ischaemic penumbra brain tissue in the MCAO/reperfusion group and MCAO group or the equivalent brain tissue in the sham group using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer’s instructions. An equal amount of total RNA was used for first-strand cDNA synthesis using an oligo-dT primer and M-myeloblastosis virus reverse transcriptase XL (Promega). The synthesised first-strand cDNA (2 µl) was used for PCR. The RT-PCR primers used to amplify FoxO3a were as follows: F: 5’-AAGGGAAGGAGCCGAGGTA-3’; R: 5’-CGACTCTGTGGCTCGAACTCT-3’. SYBR Green PCR Master Mix (ABI) was used for real-time RT-PCR. β-Actin was used as an internal reference control. The RT-PCR primers used to amplify rat β-actin were as follows: F: 5’-CGTAAAGACCTCTATGCCAACA-3’; R: 5’-AGCCACCAATCCACACAGAG-3’.
Western blot analysis
Total proteins were extracted from the cells in cell lysis buffer (50 mM Tris–HCl (pH 8.0), 120 mM NaCl, 0.5% NP-40, 1 mM PMSF), and the protein concentration was determined by the BCA method. The proteins (30 µg) were subjected to 8–10% SDS-polyacrylamide gel electrophoresis and transferred onto Hybond ECL membranes (Amersham). The membranes were incubated for 1 h at room temperature in blocking buffer (5% skimmed milk in TBS-T) and then incubated with the appropriate antibodies (rabbit anti-FoxO3a (cat. no. 12829, Cell Signalling Technology; 1 : 1000) and mouse anti-β-actin (Millipore; 1 : 10,000)) overnight at 4°C. After being washed with TBS-T, the membranes were incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibody (1 : 10,000; Sigma) for 2 h at room temperature. Western blot detection reagents (Odyssey) were used to detect proteins on the membranes. The corresponding bands were semi-quantitatively analysed by ImageJ (NIH, version 1.8.0).
Lentiviral vector-mediated gene knockdown or overexpression
FoxO3a knockdown and overexpression vectors were constructed by Hanyin Co. (Shanghai, China). Specifically, human FoxO3a or FoxO3a target sequences were separately cloned into the pMSCV-IRES-GFP plasmids for construction of FoxO3a overexpression or knockdown recombinant lentivirus vectors. The knockdown sequence of FoxO3a was: 5’-GCTCTTGGTGGATCATCAA-3’. To construct circFoxO3 overexpression (OE) plasmids, a basic sequence (flanked by HxoI and Agel) was synthesised by qRT-PCR. Primer sequence: F: 5’-ATTGTCCATGGAGACAGCCCGCCG-3’; R: 5’-GTGGGGAACTTCACTGGTGCTAAG-3’. A small spacer sequence containing two restriction enzyme sites, HindIII and SalI, was added for the insertion of the circRNA fragment. The recombinant and negative control (NC) lentiviruses (Hanyin Co. Shanghai, China) were prepared and tittered to 109 TU (transfection unit)/ml. After 48 h, the knockdown or overexpression efficiency was confirmed via quantitative real-time polymerase chain reaction (qRT-PCR). To obtain stable cell lines, the cells were seeded in six-well plates at a density of 2 × 105 cells per well. The cells were then infected with the same viral titre with 8 µg/ml Polybrene on the following day. Approximately 72 h after viral infection, the culture medium was replaced with selection medium containing 4 µg/ml puromycin for selecting cell lines with stable expression of targeted sequence. The cells were then cultured for at least 14 days. Puromycin-resistant cells were amplified in medium containing 2 µg/ml puromycin for 7 to 9 days and then transferred to a medium without puromycin.
Cell apoptosis analysis
Apoptosis was determined by detecting the translocation of phosphatidylserine to the cell surface using an annexin and DAPI apoptosis detection kit (BD Inc., USA). Rat neuroblastoma B35 cells were treated with CoCl2 (250 µmol) at the indicated time. Then, the cells were harvested, washed twice in cold PBS, and resuspended in annexin V-fluorescein isothiocyanate (FITC) and PI for 30 min in the dark. Cell apoptosis was analysed by using Cell Quest software on a FACSAria flow cytometer (BD Inc., USA). Fluorescence was detected with an excitation wavelength of 480 nm.
Statistical analysis
Data are presented as the mean ± the standard error of the mean. A Tukey test was conducted for multiple comparisons in conjunction with one-way analysis of variance. All statistical analyses were performed using SPSS for Windows v. 17.0 (SPSS, Chicago, IL). All results were considered significant with a two-sided p-value < 0.05.
Results
The increased expression of FoxO3a after H/I in vivo and in vitro
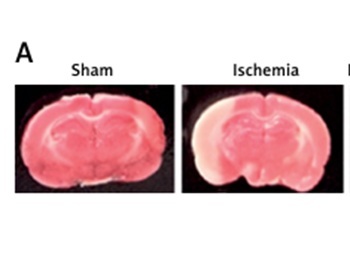
We successfully constructed a rat model of middle cerebral artery occlusion (MCAO)/reperfusion. TTC staining showed that the infarction volume was significantly increased after MCAO and even higher after reperfusion (Figures 1 A, B). Both RT-PCR and western blot revealed that FoxO3a was significantly upregulated during MCAO and decreased after reperfusion in vivo (Figures 1 C, D). The caspase-3, caspase-7 and Bax levels were also significantly increased in I/R model (Figure 1 D). Similarly, in B35 cells treated with CoCl2, the expression of FoxO3a was significantly increased after 24 h, with a peak at 48 h, and decreased at 72 h (Figures 1 E, F), indicating that the expression of FoxO3a was increased after H/I both in vivo and in vitro. The caspase-3, caspase-7, and Bax levels were significantly increased at 72 h (Figure 1 F).
Figure 1
Increased expression of FoxO3a after H/I exposure in vivo and in vitro. A – Gross examination of the infarct brain tissues in rats in the sham, MCAO, and MCAO/reperfusion groups. B – Measurements of infarct volume in three groups by TTC staining, expressed as the ratio of the infarct volume to the total volume. C – Relative expression levels of FoxO3a in the sham, MCAO, and MCAO/reperfusion groups. D – Expression levels of FoxO3a, Bax, caspase-3, and caspase-7 in the sham, MCAO, and MCAO/reperfusion groups. E – Relative FoxO3a mRNA expression levels in B35 cells treated with CoCl2 for 24 h, 48 h, and 72 h. F – Expression levels of FoxO3a, Bax, caspase-3, and caspase-7 in B35 cells treated with CoCl2 for 24 h, 48 h, and 72 h
The knockdown of FoxO3a increased apoptotic neuronal cells in response to hypoxia/ischaemia
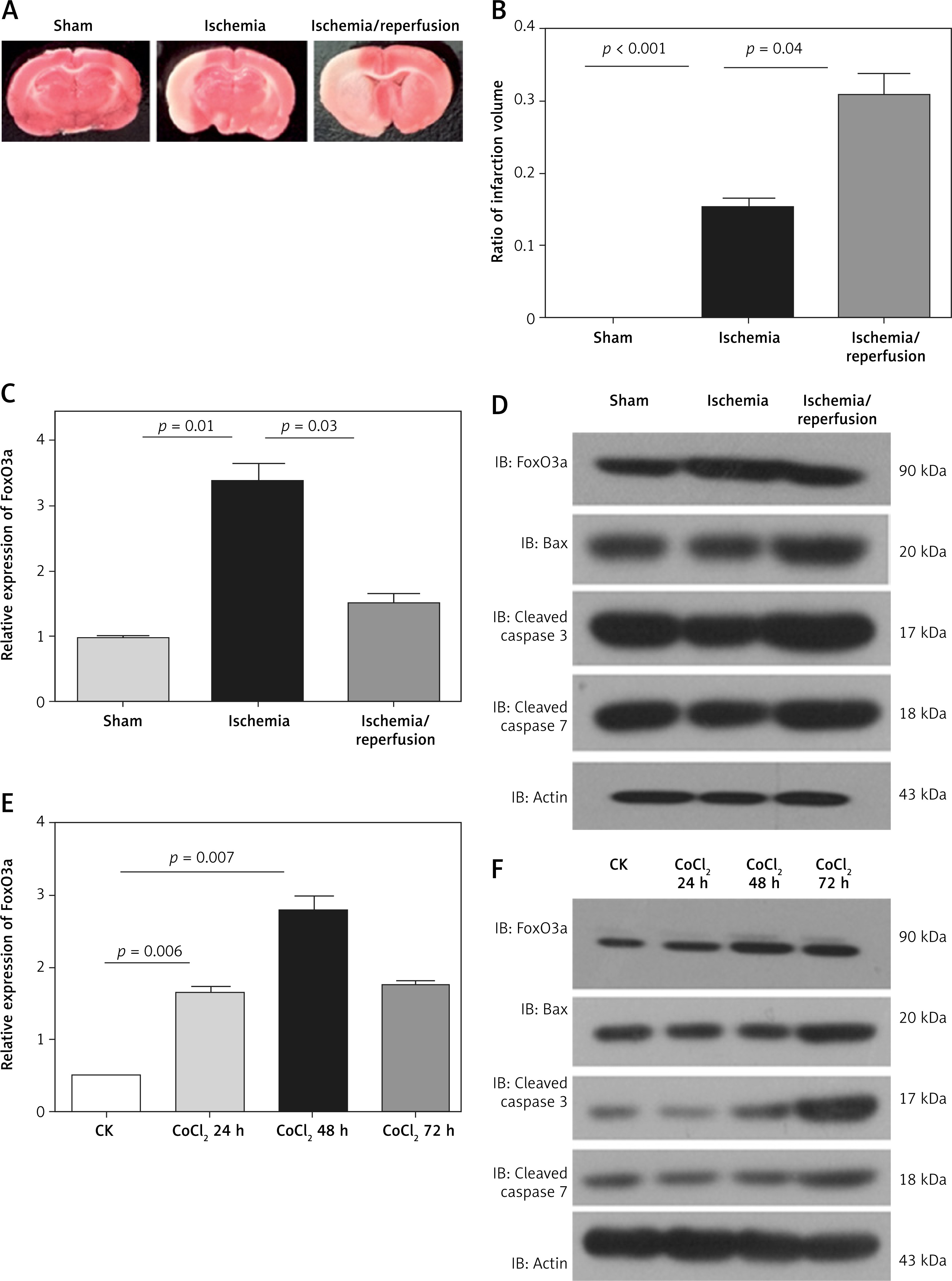
To investigate the function of FoxO3a in hypoxia/ischaemia, we constructed stable FoxO3a knockdown B35 cells using shRNA lentivirus. As shown in Figures 2 A and B, FoxO3a was efficiently inhibited in knockdown (KD) cells compared to negative control (NC) cells (Figures 2 A, B). An apoptosis assay of these cells after CoCl2 treatment showed that FoxO3a suppression increased the percentage of apoptotic cells compared to that in control cells (Figures 2 C, D). Altogether, these results indicate that the knockdown of FoxO3a in neuronal cells elevated the percentage of apoptotic cells after ischaemia and they imply a neuroprotective effect of FoxO3a.
Figure 2
The knockdown of FoxO3a expression increased apoptotic neuronal cells in response to hypoxia/ischaemia. A, B – Relative FoxO3a mRNA and protein expression levels in B35 cells with or without FoxO3a knockdown. C, D – Percentage of apoptotic B35 cells with or without FoxO3a knockdown treated with CoCl2 for 72 h. All the experiments were repeated three times
The overexpression of FoxO3a reduced apoptotic neuronal cells in response to hypoxia/ischaemia
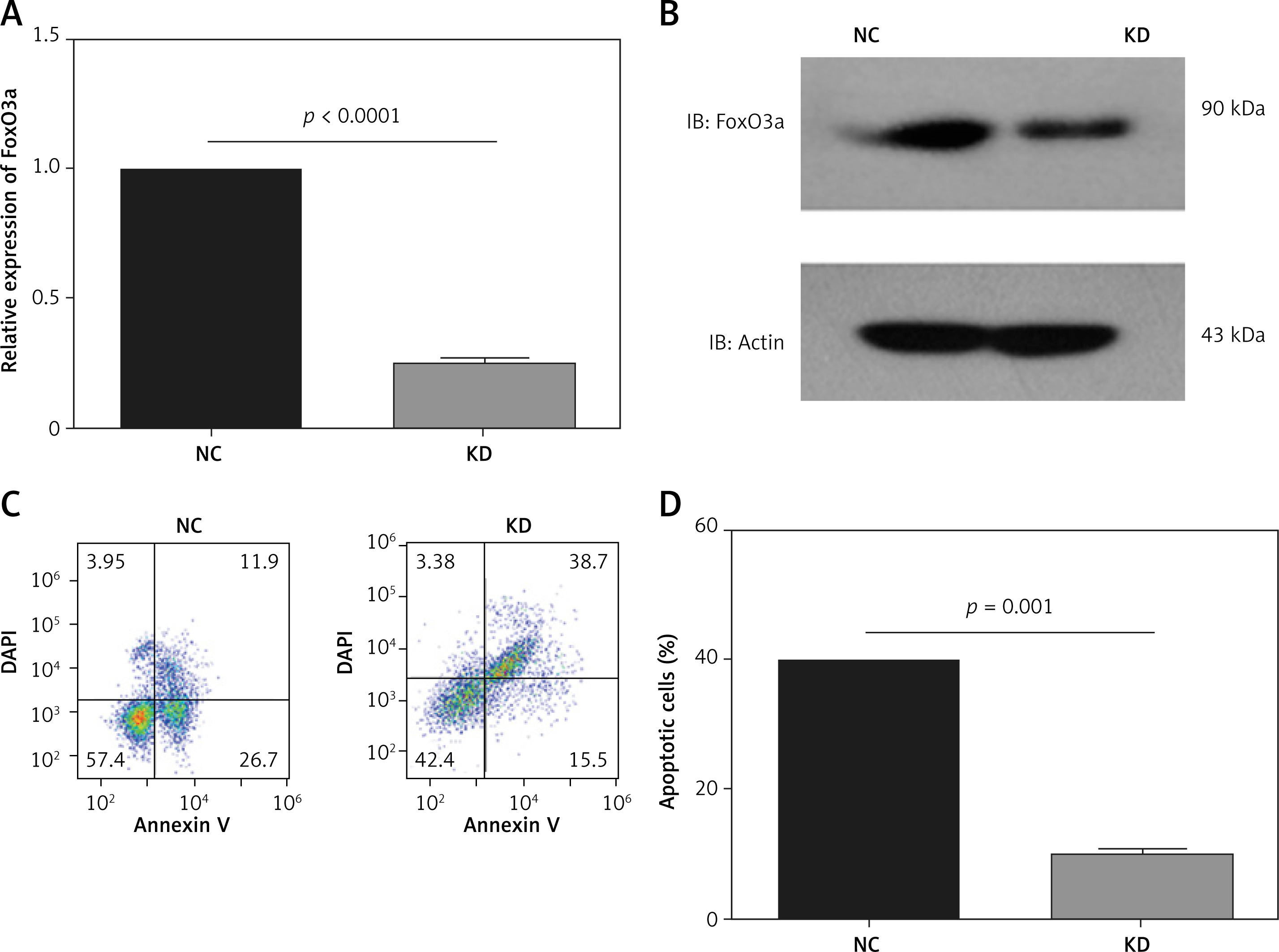
To further clarify the neuroprotective function of FoxO3a in hypoxia/ischaemia, we constructed stable FoxO3a-overexpressing B35 cells using lentivirus. As shown in Figures 3 A and B, FoxO3a was efficiently overexpressed in OE cells compared to negative control (NC) cells (Figures 3 A, B). An apoptosis assay in these cells after CoCl2 treatment showed that FoxO3a overexpression reduced the percentage of apoptotic cells (Figures 3 C, D). Altogether, these results indicate that the overexpression of FoxO3a in neuronal cells reduced the percentage of apoptotic cells after ischaemia.
Figure 3
The overexpression of FoxO3a reduced the proportion of apoptotic neuronal cells in response to hypoxia/ischaemia. A, B – Relative FoxO3a mRNA and protein expression levels in B35 cells with or without FoxO3a overexpression. C, D – Percentage of apoptotic B35 cells with or without FoxO3a overexpression treated with CoCl2 for 72 h. All the experiments were repeated three times
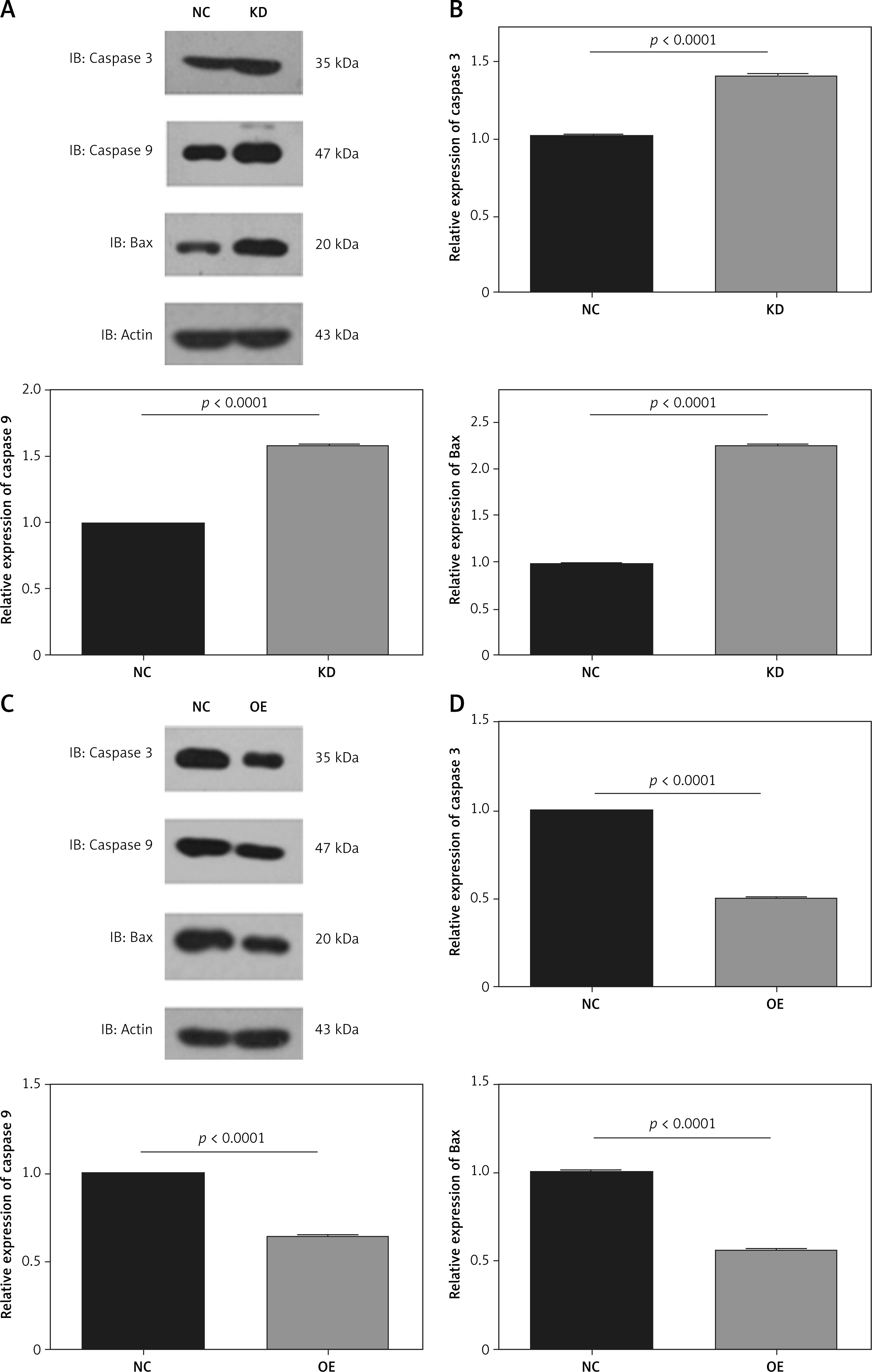
The upregulation of FoxO3a suppresses apoptotic protein levels in neuronal cells in response to hypoxia/ischaemia
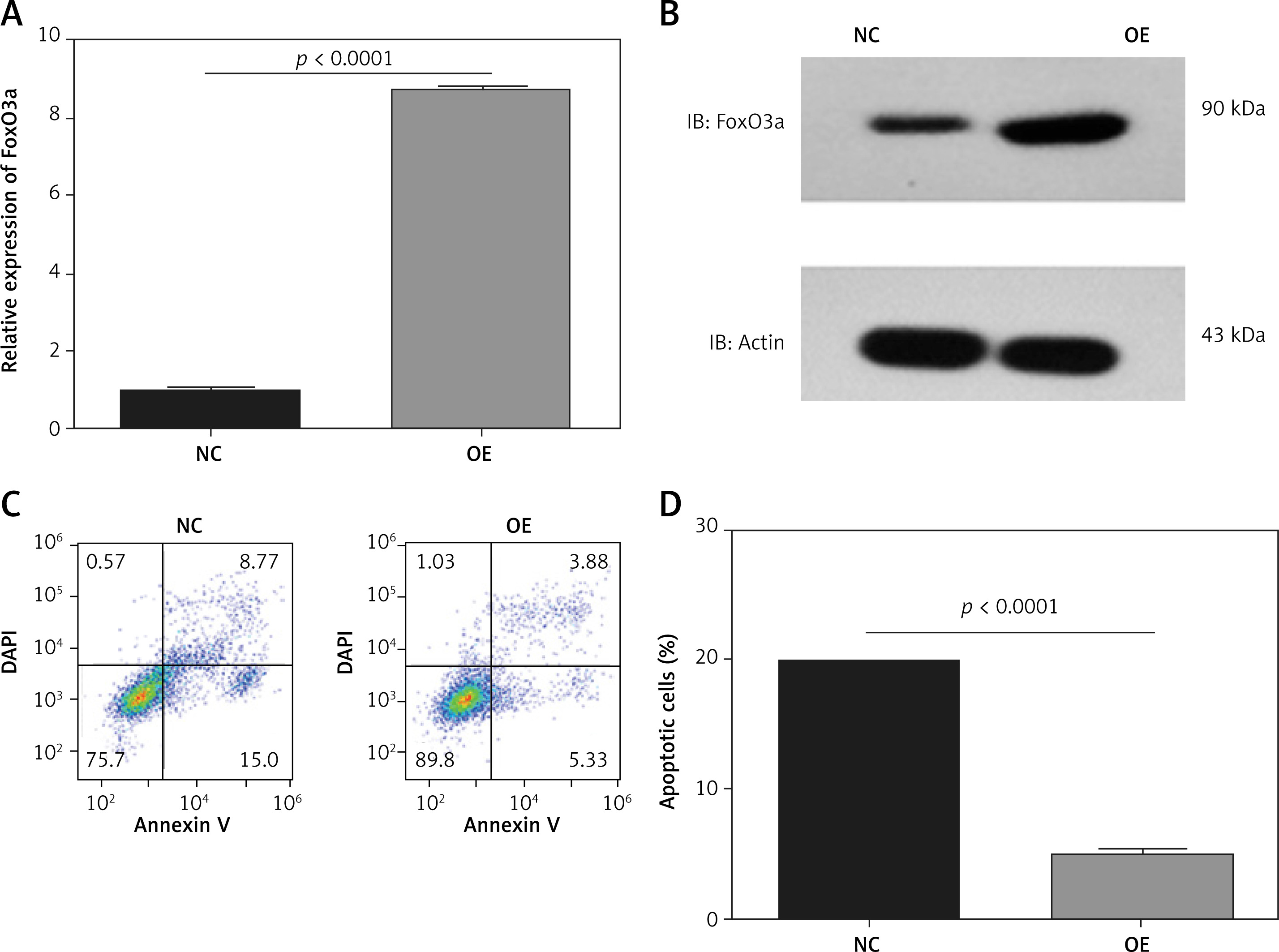
We further analysed the apoptotic proteins in FoxO3a knockdown or FoxO3a-overexpressing cells. Western blotting verified that the expression of the apoptotic proteins including caspase-3, caspase-9, and Bax were increased after FoxO3a knockdown (Figures 4 A, B). In contrast, the expression of the apoptotic marker caspase-3, caspase-9, and Bax were significantly reduced after FoxO3a overexpression (Figures 4 C, D). Altogether, these results indicate that FoxO3a protects neuronal cells from hypoxia/ischaemia-induced apoptosis partially through inhibition of the apoptotic pathway.
Figure 4
Upregulated FoxO3a expression suppressed apoptotic proteins in neuronal cells in response to hypoxia/ischaemia. A, B – Relative expression levels of caspase-3, caspase-9, and Bax in B35 cells with or without FoxO3a knockdown treated with CoCl2 for 72 h. C, D – Relative expression levels of caspase-3, caspase-9, and Bax in B35 cells with or without FoxO3a overexpression treated with CoCl2 for 72 h
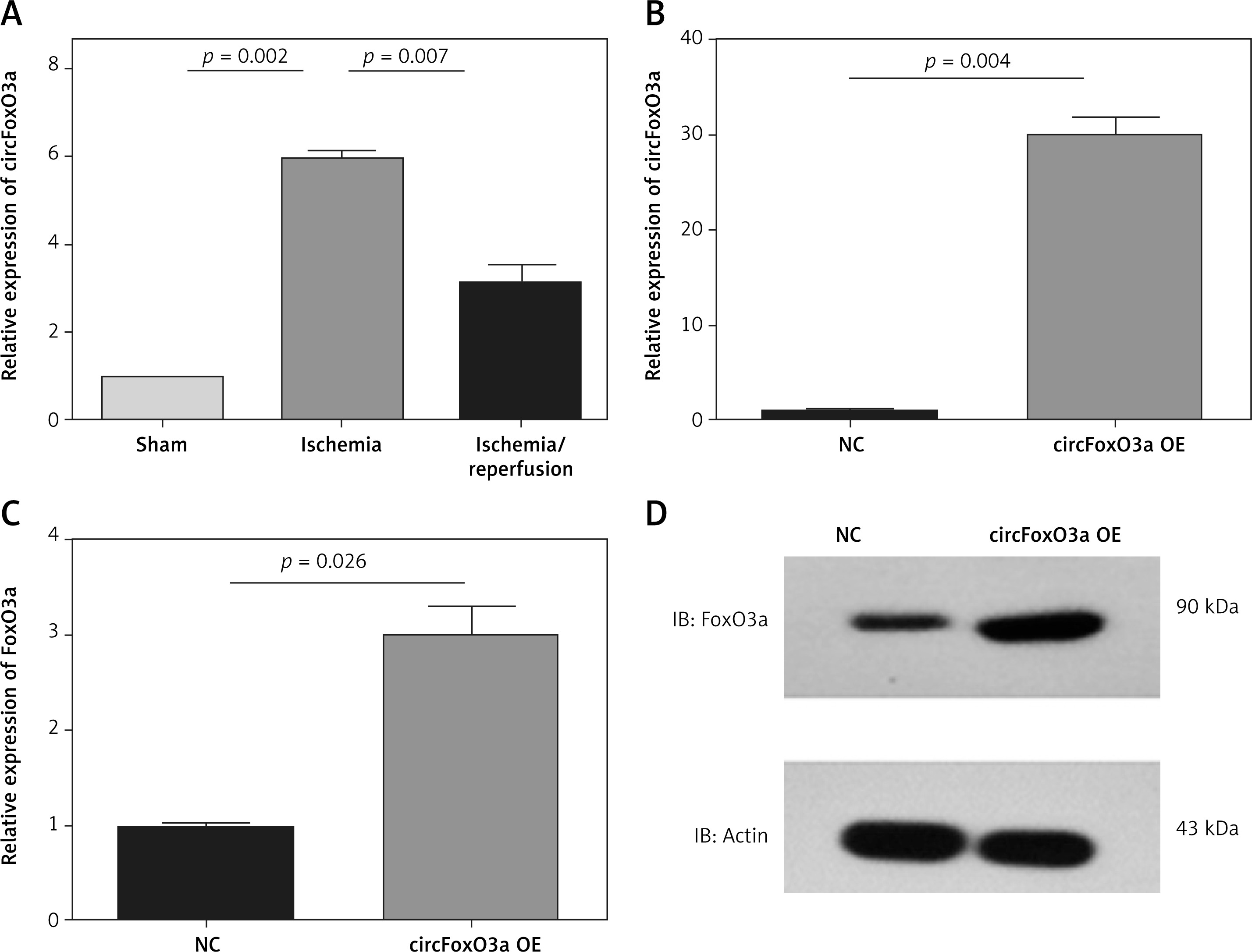
The upregulation of circFoxO3 enhanced FoxO3a expression levels in neuronal cells
We further analysed the expression of circFoxO3, and the results showed that consistent with FoxO3a, circFoxO3 was significantly upregulated during MCAO and decreased after reperfusion in vivo (Figure 5 A). We thus constructed stable circFoxO3-overexpressing B35 cells using lentivirus (Figure 5 B). Overexpression of circFoxO3 significantly enhanced expression of FoxO3a (Figures 5 C, D) indicating that the upregulation of circFoxO3 enhanced FoxO3a expression levels in neuronal cells.
Figure 5
The upregulation of circFoxO3 enhanced FoxO3a expression levels in neuronal cells. A – Relative expression levels of circFoxO3 in the sham, MCAO, and MCAO/reperfusion groups. B – Relative expression levels of circFoxO3 in the NC and circFoxO3 OE cells. C, D – Relative expression levels of FoxO3a in B35 cells with or without circFoxO3 overexpression
Discussion
In a rat model of MCAO/reperfusion and in CoCl2-treated (hypoxia mimetic) rat neuroblastoma cells, this study showed that the circRNA and protein expression of FoxO3a increased after ischaemia/hypoxia exposure. Furthermore, the suppression of FoxO3a expression in B35 cells significantly increased hypoxic cell apoptosis. In contrast, the overexpression of FoxO3a significantly reduced hypoxic cell apoptosis. As expected, circFoxO3 and FoxO3a had consistent variation trends during ischaemia/hypoxia exposure, with the underlying mechanisms to be revealed in future work.
FoxO3a was reported to exhibit stronger immunostaining in the cytoplasm of breast cancer cells than in benign breast tissues, which predicted poor survival [15]. In acute myeloid leukaemia, the viability of leukaemia stem cells depended on the expression of FoxO3a [40]. These complex observations underscore the complicated dual functions of FoxO3a [40]. In the field of tumour targeted therapy, it was proven that targeting the non-canonical AKT-FoxO3a axis turned out to be a novel therapeutic strategy for treating oral squamous cell carcinoma (OSCC) patients [41]. In experimental colon cancer, quinazoline-based small molecule suppressed tumour progression by inhibiting PI3K/Akt/FoxO3a signalling pathway [42]. In this study, we revealed the expression and superficial functions of FoxO3a in cerebral ischaemia/reperfusion. The precise signalling pathway of FoxO3a in regulating ischaemia/hypoxia-induced apoptosis needs to be explored in order to identify clinical potential of FoxO3a (or other molecules in the pathway) as a neuroprotective agent.
Several studies have shown that FoxO3a protects cells from apoptosis and promotes invasion and metastasis under stress conditions through the regulation of the nuclear factor κB (NF-κB) signalling pathway and the transcriptional regulation of genes involved in cell migration [8, 21, 43–45]. Consistently, our study revealed that upregulated FoxO3a expression protected neuronal cells from hypoxia/ischaemia-induced apoptosis via inactivation of the apoptotic pathway. However, some limitations have to be addressed. Although a relationship between FoxO3a and hypoxic apoptosis has previously been reported [29, 44], the specific mechanisms of FoxO3a in protecting neuronal cells under ischaemia/hypoxia stress still need further investigation.
The mechanism underlying the biological function of circFoxO3 is currently unknown. As shown in previous studies, circRNAs may act as endogenous sponges to interact with miRNAs and therefore regulate the expression of miRNA target genes. For instance, circRNA TLK1 functions as a miR-355 sponge to aggravate neuronal injury and neurological deficits after ischaemic stroke [46]. It is worth noting that circFoxO3 exerts pro-tumourigenic activity in glioblastoma by sponging miR-138-5p /miR-432-5p, which promotes expression of target gene named nuclear factor of activated T cells (NFAT5) [36]. In accordance with these findings, there is a possibility that circFoxO3 also functions as miRNA sponge to regulate neuronal injury during ischaemia/hypoxia. To prove this hypothesis, deep database mining and sequence alignment analysis may be optional strategies for discovering potential miRNA targets of circFoxO3. The interaction between circFoxO3 and FoxO3a, which have consistently changing trends during cerebral ischaemia/hypoxia, needs to be verified in future studies as well.
Because there are several signalling pathways involved in the rat model of ischemia/reperfusion injury [47], especially the neuroinflammation [48] and PI3K-Akt pathway [47, 49–51], the effect of circFoxO3 and FoxO3a might have an effect also on other pathways. Therefore, future studies are needed to elucidate the specific role of FoxO3a.
In conclusion, our study is the first to establish that upregulated FoxO3a expression protects neuronal cells from hypoxia/ischaemia-induced apoptosis via activation of the apoptotic pathway. The therapeutic potential of FoxO3a and (or) circFoxO3 as neuroprotective agents is worth exploring.