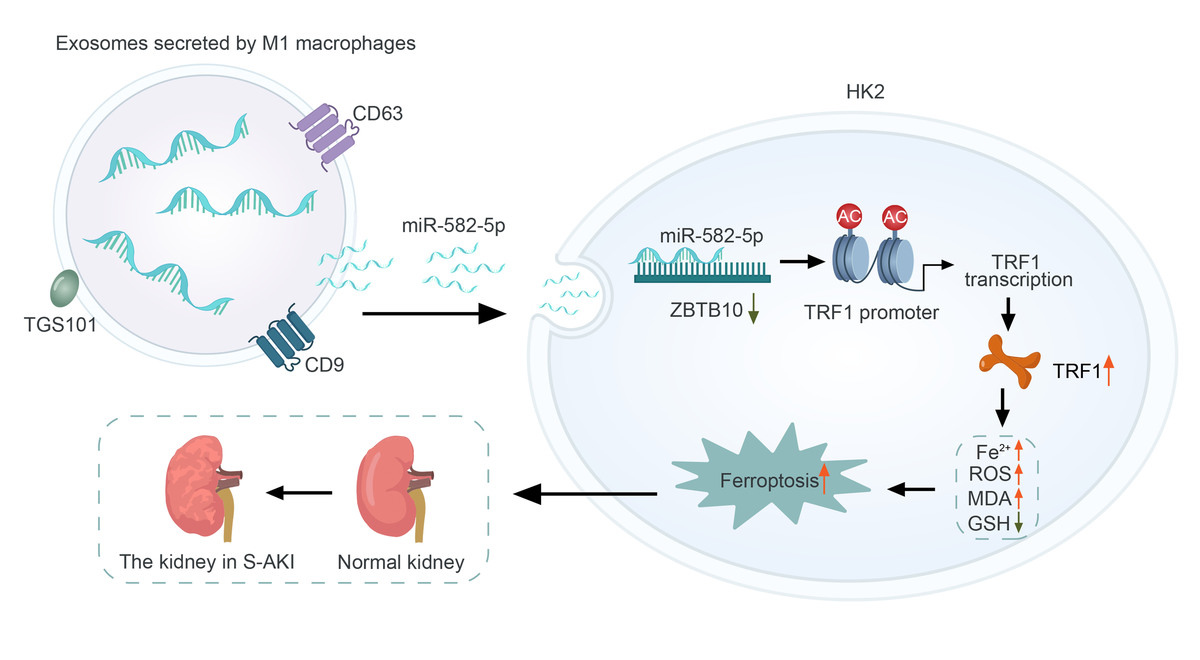
Introduction
Sepsis-induced acute kidney injury (S-AKI) is a critical condition characterized by a sudden decline in renal function [1], significantly contributing to the high mortality rates observed in septic patients [2]. The pathogenesis of S-AKI is complex and multifactorial, involving hemodynamic changes, systemic inflammation, oxidative stress, and various forms of cell death [3, 4]. Among these, ferroptosis – an iron-dependent form of regulated cell death characterized by the accumulation of lipid peroxides – has recently garnered attention for its role in AKI [5, 6]. A deeper understanding of the mechanisms leading to ferroptosis in S-AKI is crucial for identifying potential therapeutic targets.
Macrophages are key players in the innate immune response and exhibit remarkable plasticity, allowing them to adopt different functional states. M1-polarized macrophages, which exhibit a pro-inflammatory phenotype [7], secrete a variety of bioactive molecules, including cytokines, chemokines, and extracellular vesicles such as exosomes. Exosomes are small, membrane-bound vesicles that facilitate intercellular communication by transferring proteins, lipids, and nucleic acids to recipient cells [8, 9]. This intercellular communication can profoundly influence cellular behavior and fate [10, 11]. Despite extensive research on exosome biology, the specific contributions of M1 macrophage-derived exosomes to the pathogenesis of S-AKI remain underexplored.
MicroRNAs (miRNAs) are small, non-coding RNAs that regulate gene expression at the post-transcriptional level by binding to the 3′ untranslated regions (UTRs) of target mRNAs [12, 13], leading to their degradation or translational repression. MiRNAs are involved in a wide array of biological processes [14, 15], including cell proliferation, differentiation, and apoptosis, and have recently been implicated in the regulation of ferroptosis [16]. Among them, miR-582-5p has been identified as a significant regulator in various pathological contexts [17, 18], influencing cell proliferation and death. However, the role of miR-582-5p in renal tubular epithelial cells (HK2) and its potential contribution to S-AKI has not been fully elucidated.
This study aims to investigate the role of exosomes derived from M1-polarized macrophages in the proliferation and ferroptosis of renal tubular epithelial cells. We hypothesize that these exosomes carry bioactive molecules, including miR-582-5p, which modulate the viability and oxidative stress responses of HK2 cells, thereby promoting ferroptosis. By exploring the molecular mechanisms through which miR-582-5p influences the expression of ferroptosis-related genes, we sought to uncover novel insights into the pathogenesis of S-AKI and identify potential therapeutic targets for intervention.
Understanding the interactions between M1 macrophage exosomes and renal tubular epithelial cells in the context of S-AKI could provide new perspectives on the molecular pathways involved. By focusing on the role of miR-582-5p and its regulatory effects on ferroptosis, this research aimed to highlight potential strategies for preventing or mitigating kidney damage in sepsis patients.
Material and methods
Cell culture
HK2, THP-1, and RAW264.7 cells were purchased from Saibekang (Shanghai) Biotechnology Co., Ltd. HK2 cells were cultured in MEM (with NEAA) supplemented with 10% fetal bovine serum. THP-1 cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 0.5 mM β-ME. RAW264.7 cells were cultured in high-glucose DMEM supplemented with 10% fetal bovine serum.
To knock down miR-582-5p, HK2 cells were transfected with 100 nM miRNA inhibitor or NC inhibitor using Lipofectamine 3000. For overexpression of miR-582-5p, HK2 cells were transfected with 100 nM miRNA mimics or NC mimics using Lipofectamine 3000. Media were changed 6 h after transfection, followed by treatment with exosomes for 48 h before cell collection for subsequent assays. For ZBTB10 overexpression, cells were co-transfected with 4 µg of ZBTB10 overexpression plasmid or control plasmid, and cells were collected 48 h after transfection for downstream experiments.
Induction of macrophage M1 polarization and exosome isolation
RAW264.7 cells were treated with 100 ng/ml lipopolysaccharide (LPS) and 10 ng/ml IFN-γ to induce M1 polarization. THP-1 cells were treated with 100 ng/ml phorbol 12-myristate 13-acetate (PMA) for 48 h to differentiate into M0 macrophages, followed by treatment with 100 ng/ml LPS and 10 ng/ml IFN-γ for M1 polarization. Well-growing RAW264.7 and THP-1 cells were plated on 10 cm dishes at 70% confluence or 2 × 107 cells/dish, respectively, with three dishes prepared for each. After M1 polarization, the medium containing 10% fetal bovine serum was replaced with serum-free medium, and the cells were cultured for 16 h. The supernatant was collected, filtered through a 0.22 µm filter, and concentrated to approximately 700 µl using an Amicon Ultra (AU) ultrafiltration tube (Millipore), yielding concentrated crude exosomes.
Exosome tracing experiment
Concentrated crude exosomes were labeled with the red lipophilic dye PKH26 (Sigma, MINI26). PKH26 was diluted to a final concentration of 5 µM with Diluent C and incubated at room temperature for 5 min. Labeled exosomes were centrifuged at 100,000 × g for 70 min, washed three times with PBS, and resuspended in HK2 cell culture medium. Labeled exosomes were added to HK2 cells (20 µg/ml) and incubated for 16 h. Cells were stained with 1 × Hoechst 33342 to label nuclei and observed under an inverted fluorescence microscope.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted using the kit (Tiangen, DP419) according to the manufacturer’s instructions. cDNA synthesis was performed using the HiScript II Q Select RT SuperMix for qPCR (+gDNA wiper) kit (Vazyme, R233). Gene expression was analyzed using ChamQ Blue Universal SYBR qPCR Master Mix (Vazyme, Q312). Primer sequences are listed in the attached primer list.
Western blot
Protein extraction was performed using a protein extraction kit (Proteintech, PK10020). Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Beyotime, P0012AC) according to molecular weight and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore). Membranes were blocked with 5% non-fat milk in tris-buffered saline Tween-20 (TBST) and incubated with primary antibodies at 37°C for 1 h, followed by four washes with TBST. Membranes were then incubated with corresponding secondary antibodies at 37°C for 1 h, followed by four washes with TBST. Detection was performed using ECL reagent and chemiluminescence imaging. Antibodies used included GPX4 (Proteintech, 67763-1-Ig, 1 : 5000), NRF2 (ABclonal, AP1133, 1 : 2000), ACSL4 (ABclonal, A20414, 1 : 5000), TFR1 (ABclonal, A5865, 1 : 1000), ZBTB10 (Proteintech, 22944-1-AP, 1 : 300), CD63 (Proteintech, 25682-1-AP, 1 : 1000), CD9 (Proteintech, 20597-1-AP, 1 : 3000), TSG101 (Proteintech, 28283-1-AP, 1 : 8000), β-actin (Proteintech, 81115-1-RR, 1 : 20000), and GAPDH (Proteintech, 60004-1-Ig, 1 : 200000).
Cell Counting Kit-8 (CCK-8)
Cell proliferation was assessed using Cell Counting Kit-8 (Dojindo, CK04). Cells (104) were seeded into 96-well plates. CCK-8 solution (10 µl) was added at 0 h, 24 h, 48 h, and 96 h, and incubated for 2 h before measuring absorbance at 450 nm using an automated microplate reader (BioTek, 800-TS).
5-Ethynyl-2′deoxyuridine (EdU) DNA synthesis and cell proliferation were assessed using the EdU Detection Kit (C0017S). Treated HK2 cells (104) were seeded into 96-well plates and incubated overnight. The next day, cells were incubated with 10 µM EdU working solution for 2 h, fixed with 4% paraformaldehyde at room temperature for 30 min, permeabilized with 0.5% Triton X-100 for 10 min, and incubated with Click reaction solution. Cells were counterstained with 1× Hoechst 33342 and observed under a fluorescence microscope.
Malondialdehyde (MDA) level detection
MDA levels in HK2 cells were measured using the MDA Assay Kit (Beyotime, S0131S) according to the manufacturer’s instructions.
Glutathione (GSH) level detection
GSH levels in HK2 cells were measured using the Reduced Glutathione Assay Kit (Nanjing Jiancheng, A006-1-1) according to the manufacturer’s instructions.
Fe2+ level detection
Fe2+ levels in HK2 cells were measured using the Iron Assay Kit (Abcam, Cambridge, MA, USA, ab83366) according to the manufacturer’s instructions.
Reactive oxygen species (ROS) fluorescence
Intracellular ROS levels were detected using H2DCFDA (MCE, HY-D0940). Treated HK2 cells in the logarithmic growth phase were seeded into 24-well plates at a density of 5 × 105 cells/well and incubated overnight. Cells were incubated with 10 µM DCFH-DA at 37°C for 30 min in the dark and counterstained with 1× Hoechst 33342. Intracellular DCF fluorescence was observed under a fluorescence microscope.
Transmission electron microscopy (TEM)
The HK-2 cells were initially fixed using 2% glutaraldehyde for 12 h, followed by PBS washes. Subsequently, the cells were fixed with 1% samarium tetroxide for 2 h. After fixation, the samples underwent dehydration with a graded ethanol series and were stained with toluidine blue. Mitochondrial morphology was then examined using TEM.
RNA immunoprecipitation (RIP)
RIP assays were performed using the Magna RIP Kit (Millipore, 17-700) in HK2 cells. Cell lysates (over 2 × 107 cells/IP) were incubated with magnetic beads pre-coated with 5 µg Ago2 antibody (CST, 2897) or rabbit IgG (Millipore) at 4°C overnight. After washing, the RNA-protein complexes were eluted and RNA was purified using column-based extraction. Target RNA enrichment was assessed by RT-qPCR.
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed using the Magna ChIP A/G kit (Millipore, 17-10085) in HK2 cells. Fixed cells (5 × 106 cells/IP) were lysed using Cell Lysis Buffer, and nuclei were resuspended in Nuclear Lysis Buffer. DNA was sheared to 200–1000 bp fragments by sonication (150 W, 6 s on, 30 s off, 25 cycles). Chromatin was incubated with 5 µg of antibody at 4 °C overnight, with rabbit IgG as a negative control and 10% input as a positive control. DNA was eluted and purified, and target DNA fragments were assessed by qPCR.
Pull down
HK2 cells (1 × 107) were lysed with RIP lysis buffer. Bio-NC or Bio-miR-582-5p probes were incubated with C1 magnetic beads (Life) at 25°C for 2 h to generate probe-coated beads, which were then incubated with cell lysates at 4°C overnight. After washing, the RNA mixture was eluted and purified using the SteadyPure RNA Extraction Kit (AG, AG21024), and enrichment was assessed by RT-qPCR. Bio-NC sequence: Bio-5′-TTTTTTTTTTTTTTTTTTTT-3′; Bio-miR-582-5p sequence: Bio-5′-TTACAGTTGTTCAACCAGTTACT-3′.
Luciferase reporter assay
The TFR1 promoter region was cloned into the pGL3 basic vector, and HK2 cells were co-transfected with reporter plasmid and shNC, shZBTB10#1, or shZBTB10#2 using Lipofectamine 3000, with pRL-TK as a transfection control. Dual luciferase activity was measured using the Dual Luciferase Reporter Assay Kit (Vazyme, DL101-01). Relative luciferase activity was calculated as the ratio of firefly to Renilla luciferase activity.
The ZBTB10 3′UTR regions were cloned into the pmir-GLO vector, and HK2 cells were co-transfected with NC mimics or miR-582-5p mimics and the reporter plasmid using Lipofectamine 3000. Dual luciferase activity was measured using the Dual Luciferase Reporter Assay Kit (Vazyme, DL101-01). Relative luciferase activity was calculated as the ratio of Renilla to firefly luciferase activity.
Statistical analysis
Data are presented as mean ± standard deviation (SD) from at least three independent experiments. Statistical analyses were performed using GraphPad Prism software. Differences between groups were assessed using Student’s t-test or one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test for multiple comparisons. The Shapiro-Wilk test was used to assess the normality of the data, and after confirming that the data followed a normal distribution, ANOVA was performed. A p-value of < 0.05 was considered statistically significant.
Results
M1-polarized macrophage exosomes inhibit proliferation and promote ferroptosis in renal tubular epithelial cells (HK2)
To investigate the mechanism by which M1 macrophages, through the exosomal pathway, affect AKI induced by sepsis, we polarized THP-1 and RAW264.7 cells to the M1 phenotype and confirmed the M1 polarization markers via RT-qPCR (Supplementary Figure S1 A). After isolating exosomes from M1-polarized macrophages and analyzing their purity using Western blot (Supplementary Figure S1 B), we treated HK2 cells with these exosomes (Supplementary Figure S1 C). Exosome tracing experiments revealed that M1-polarized macrophages could communicate with HK2 cells via the exosomal pathway (Supplementary Figure S1 D).
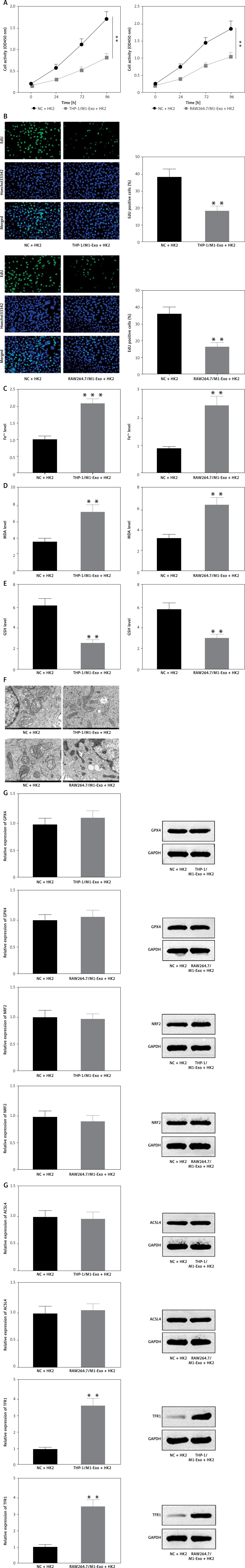
Following treatment with M1-polarized macrophage exosomes, CCK-8 and EdU assays showed that the proliferative capacity of HK2 cells was significantly reduced (Figure 1 A, B). This confirmed the inhibitory effect of M1-polarized macrophage exosomes on the proliferation of HK2 cells. Given that ferroptosis plays a critical role in the development of cardiovascular diseases, particularly in sepsis-induced cardiomyopathy, precise detection of ferroptosis is crucial for early intervention and treatment [19]. Therefore, we conducted a series of experiments to assess the level of ferroptosis in HK2 cells. Compared to the control group, the levels of Fe2+ and MDA were significantly elevated in HK2 cells treated with exosomes, while the level of GSH was markedly reduced (Figures 1 C–E), indicating the occurrence of ferroptosis. To further confirm these phenotypic changes, we used TEM to observe the morphological changes of mitochondria in HK2 cells under different treatments. We found that the mitochondrial volume in exosome-treated cells was reduced, and the mitochondrial cristae were diminished or absent, exhibiting morphological hallmarks of ferroptosis (Figure 1 F). Additionally, we assessed the mRNA and protein levels of ferroptosis-inhibiting genes (NRF2, GPX4) and ferroptosis-promoting genes (ACSL4, TFR1) via RT-qPCR and Western blot (Figure 1 G). The results showed that TFR1 expression was significantly upregulated at both mRNA and protein levels following exosome treatment, while other genes did not exhibit significant changes at either level. This suggests that M1-polarized macrophage exosomes may induce ferroptosis in HK2 cells by promoting TFR1 expression.
Figure 1
M1-polarized macrophage exosomes inhibit proliferation and promote ferroptosis in renal tubular epithelial cells (HK2). Treatment of HK2 cells with M1-polarized macrophage exosomes impacts HK2 cell phenotype. Groups: NC+HK2, THP-1/M1-Exo+HK2; and NC+HK2, RAW264.7/M1-Exo+HK2. A, B – CCK-8 and EdU assays to detect HK2 cell proliferation under different treatment conditions. C – Measurement of Fe2+ levels in HK2 cells under different treatment conditions. D – Measurement of MDA levels in HK2 cells under different treatment conditions. E – Measurement of GSH levels in HK2 cells under different treatment conditions. F – TEM was used to observe the differences in mitochondrial morphology of HK2 cells under different treatment conditions. G – RT-qPCR and Western blot analysis of ferroptosis-related genes (NRF2, GPX4, ACSL4, and TFR1) in HK2 cells under different treatment conditions
MiR-582-5p inhibits HK2 proliferation and promotes ferroptosis
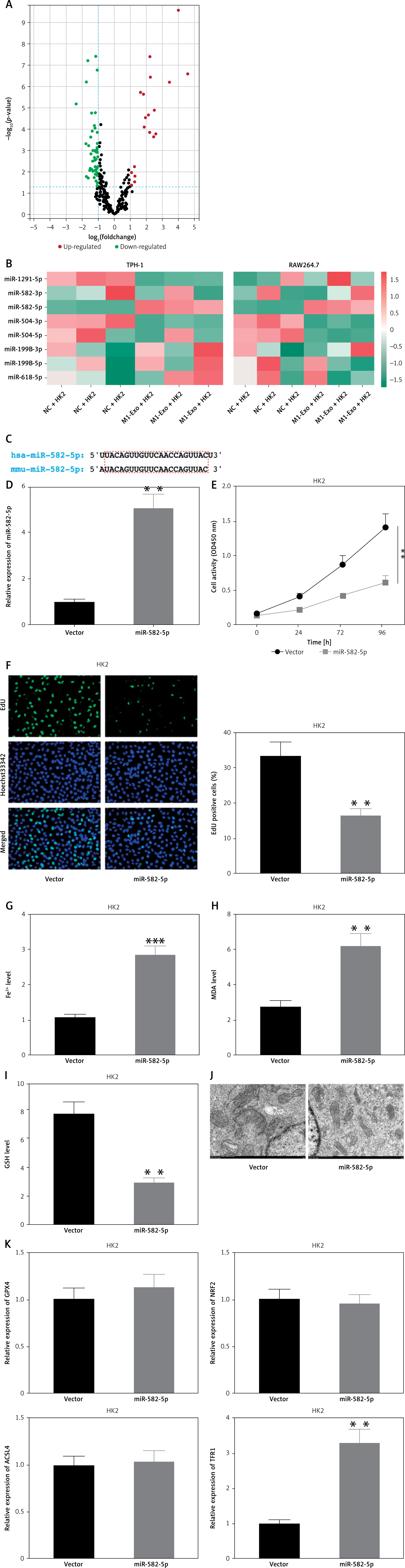
MiRNAs play a crucial role in molecular dynamics mediated by exosomes during sepsis [20]. By analyzing the GEO database series GSE242059, we selected the top five significantly upregulated miRNAs as candidate genes (Figure 2 A). To determine whether the regulatory effect of miRNAs is mediated through the exosomal pathway, we used RT-qPCR to measure the expression changes of candidate genes in HK2 cells (Figure 2 B). The results indicated that miR-582-5p expression was significantly upregulated in HK2 cells treated with exosomes derived from RAW264.7 and THP-1 cells. This finding drew our attention, and further analysis confirmed the high species conservation of the miR-582-5p sequence (Figure 2 C), suggesting its critical role in regulating gene expression, developmental processes, and disease mechanisms. Subsequently, we overexpressed miR-582-5p (Figure 2 D), and the CCK-8 and EdU results showed that HK2 cell proliferation was inhibited (Figures 2 E, F). Fe2+ and MDA levels increased, and GSH levels significantly decreased due to miR-582-5p overexpression (Figures 2 G–I). The TEM images also indicated that overexpression of miR-582-5p resulted in ferroptotic morphology in HK2 cells (Figure 2 J). RT-qPCR and Western blot confirmed that miR-582-5p promoted TFR1 expression in a manner consistent with the effects observed in HK2 cells treated with M1-polarized macrophage-derived exosomes, while NRF2, GPX4, and ACSL4 were unaffected (Figure 2 K).
Figure 2
MiR-582-5p inhibits HK2 proliferation and promotes ferroptosis. A – Differential gene screening results from GSE242059. B – RT-qPCR analysis of candidate miRNA expression in M1-polarized macrophages. C – Conservation sequence of miR-582-5p. Effect of miR-582-5p overexpression on HK2 cells. Groups: vector, miR-582-5p. D – RT-qPCR analysis of miR-582-5p expression in different groups. E, F – CCK-8 and EdU assays to detect HK2 cell proliferation in different groups. G – Measurement of Fe2+ levels in HK2 cells in different groups. H – Measurement of MDA levels in HK2 cells in different groups. I – Measurement of GSH levels in HK2 cells in different groups. J – TEM was used to observe the differences in mitochondrial morphology of HK2 cells in different groups. K – RT-qPCR analysis of ferroptosis-related genes (NRF2, GPX4, ACSL4, and TFR1) in HK2 cells in different groups
M1-polarized macrophage exosomes promote ferroptosis in HK2 cells via miR-582-5p
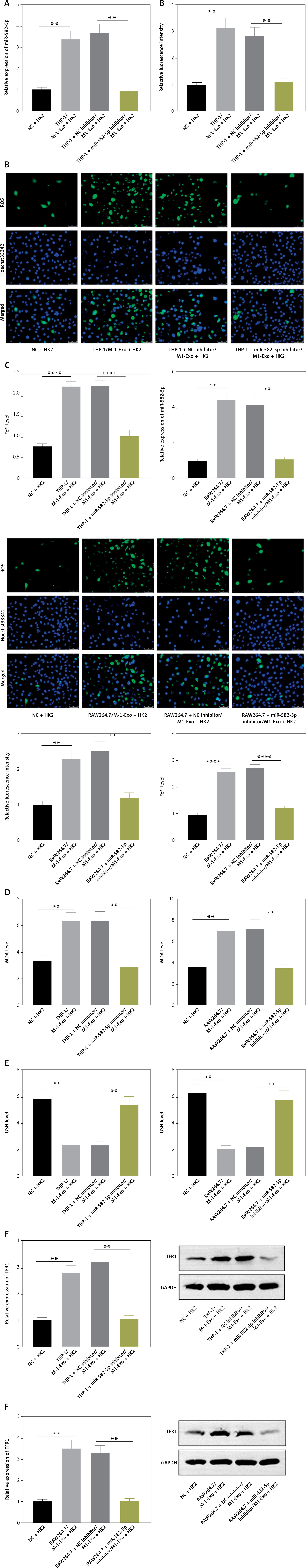
MiR-582-5p is highly expressed in M1-polarized macrophage exosomes and can promote ferroptosis in HK2 cells. Based on this, we hypothesized that M1-polarized macrophages secrete exosomes to deliver miR-582-5p to HK2 cells, thereby inducing ferroptosis. The earlier results showed high miR-582-5p expression in M1-polarized macrophage exosomes. To confirm this, we inhibited miR-582-5p in macrophages, extracted exosomes, and used them to treat HK2 cells. We then measured miR-582-5p expression in HK2 cells to determine whether the expression difference was mediated by macrophage exosomes, leading to phenotypic changes. RT-qPCR results showed that exosomes from non-inhibited macrophages led to upregulation of miR-582-5p in HK2 cells, whereas exosomes from miR-582-5p-inhibited macrophages did not (Figure 3 A). This indicates that macrophages deliver miR-582-5p to HK2 cells via exosomes, leading to abnormal miR-582-5p expression in HK2 cells. To further confirm the impact of exosome-derived miR-582-5p on HK2 cell phenotype, we measured ROS levels in similarly treated HK2 cells. Treatment with exosomes derived from M1-polarized macrophage increased ROS levels in HK2 cells, which was reversed when miR-582-5p was absent (Figure 3 B). Fe2+ and MDA levels showed similar trends (Figures 3 C, D), while GSH levels exhibited opposite changes (Figure 3 E). Additionally, the mRNA and protein levels of the ferroptosis-promoting gene TFR1 increased with the presence of exosomal miR-582-5p and were suppressed in its absence (Figure 3 F). These results indicate that M1-polarized macrophages deliver miR-582-5p to HK2 cells via exosomes, and the upregulation of miR-582-5p promotes ferroptosis in HK2 cells, leading to phenotypic changes.
Figure 3
M1-polarized macrophage exosomes promote ferroptosis in HK2 cells via miR-582-5p. Effect of miR-5825p interference in M1-polarized macrophage exosomes on HK2 cells. Groups: NC + HK2, THP-1/M1-Exo + HK2, THP-1 + NC inhibitor/M1-Exo + HK2, THP-1 + miR-582-5p inhibitor/M1-Exo + HK2; and NC + HK2, RAW264.7/M1-Exo + HK2, RAW264.7 + NC inhibitor/M1-Exo + HK2, RAW264.7 + miR-582-5p inhibitor/M1-Exo + HK2. A – RT-qPCR analysis of miR-582-5p expression in different groups. B – Measurement of ROS levels in HK2 cells in different groups. C – Measurement of Fe2+ levels in HK2 cells in different groups. D – Measurement of MDA levels in HK2 cells in different groups. E – Measurement of GSH levels in HK2 cells in different groups. F – RT-qPCR and Western blot analysis of the ferroptosis-promoting gene TFR1 in HK2 cells in different groups
MiR-582-5p binds to the 3′ UTR of ZBTB10, inhibiting ZBTB10 expression and activating ferroptosis in HK2 cells
We predicted the potential targets of miR-582-5p to analyze the mechanism through which miR-582-5p regulates HK2 cell phenotypic changes. Using the online tool ENCORI (https://rnasysu.com/encori/index.php), we predicted two potential target genes: MAP3K7 and ZBTB10 (Figure 4 A). Following miR-582-5p overexpression, RT-qPCR results showed that ZBTB10 expression was suppressed (Figure 4 B). Under the same conditions, Western blot analysis confirmed that ZBTB10 protein levels were also downregulated (Figure 4 C). This suggests that miR-582-5p may regulate ZBTB10 expression. To further confirm the binding relationship between miR-582-5p and ZBTB10, we identified five potential binding sites on the ZBTB10 3′ UTR based on the seed region (Figure 4 D). RIP experiments showed that miR-582-5p binds to the ZBTB10 3′ UTR, with significant enrichment at site 1 (Figure 4 E). This was further confirmed by luciferase reporter assays demonstrating miR-582-5p binding at site-1 of the ZBTB10 3′ UTR (Figure 4 F). MiR-582-5p binding to the ZBTB10 3′ UTR downregulated ZBTB10. Treatment of HK2 cells with M1-polarized macrophage exosomes increased miR-582-5p levels in HK2 cells, resulting in decreased ZBTB10 expression, as confirmed by RT-qPCR and Western blot (Figures 4 G, H).
Figure 4
MiR-582-5p binds to the 3′ UTR of ZBTB10, inhibiting ZBTB10 expression and activating ferroptosis in HK2 cells. A – Prediction of potential miR-582-5p targets using the online tool ENCORI. B – RT-qPCR analysis of mRNA levels of potential targets MAP3K7 and ZBTB10 in HK2 cells after miR-582-5p overexpression. C – Western blot analysis of ZBTB10 protein levels in HK2 cells after miR-582-5p overexpression. D – Potential binding sites of miR-582-5p on the ZBTB10 3′ UTR. E – RIP assay to detect the enrichment level of RISC at five target sites. F – Luciferase reporter assay to verify miR-582-5p binding at site-1 of the ZBTB10 3′ UTR. G – RT-qPCR analysis of ZBTB10 mRNA levels in HK2 cells treated with exosomes from different sources. H – Western blot analysis of ZBTB10 protein levels in HK2 cells treated with exosomes from different sources
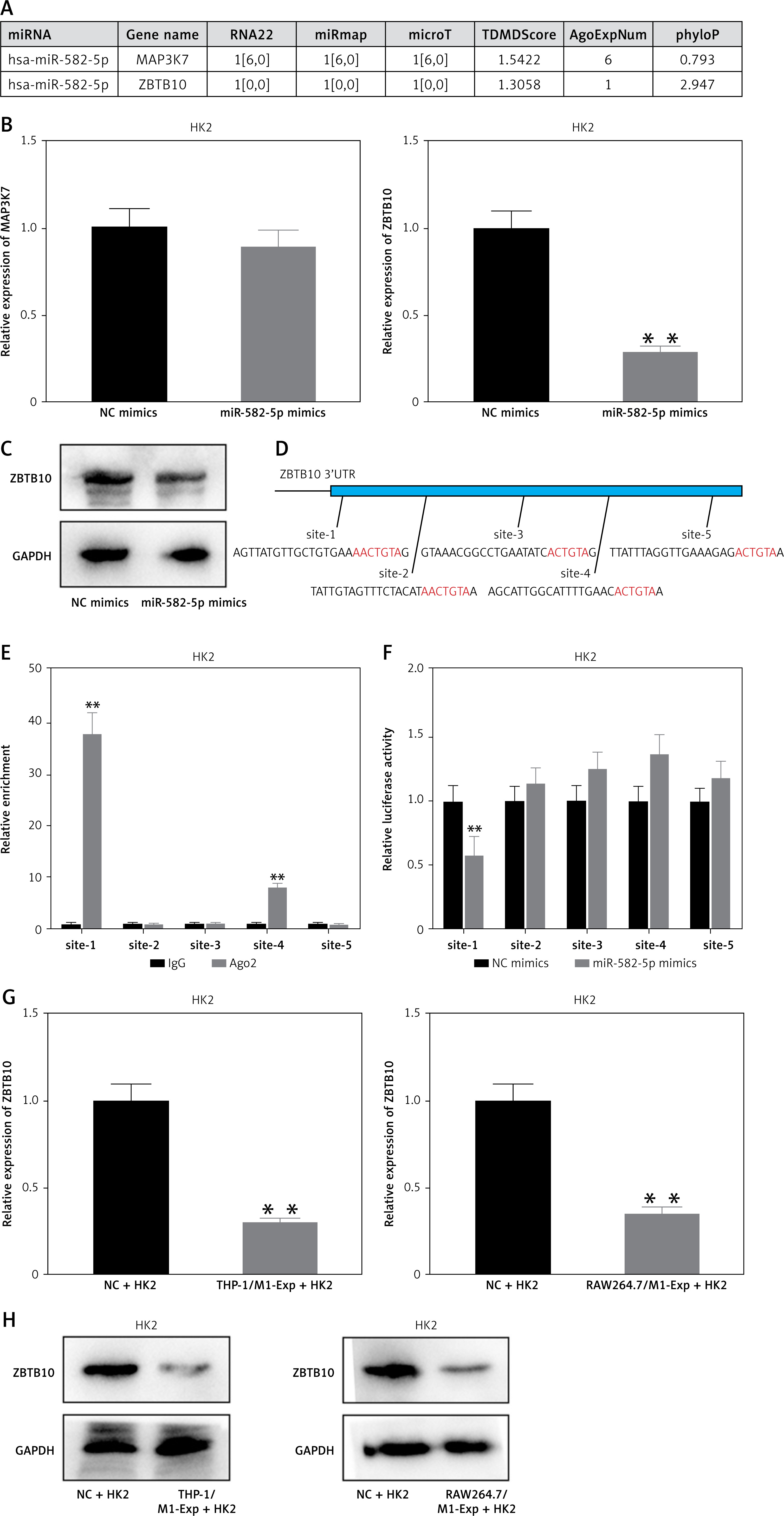
After exosome-mediated delivery of miR-582-5p to HK2 cells, ZBTB10 expression was inhibited. Further investigation is needed to determine whether ZBTB10 activation affects ferroptosis in HK2 cells. We overexpressed miR-582-5p alone or simultaneously with ZBTB10 in HK2 cells and observed changes in ferroptosis-related characteristics to confirm the role of ZBTB10 in HK2 cell ferroptosis. The results showed that miR-582-5p increased ROS, Fe2+, and MDA levels and decreased GSH levels, and these effects were reversed by overexpressing ZBTB10 (Supplementary Figures S2 A–D). The regulatory effect of miR-582-5p on TFR1 was also significantly reduced by ZBTB10 overexpression (Supplementary Figure S2 E). Therefore, miR-582-5p induces ferroptosis in HK2 cells by inhibiting ZBTB10.
Downregulation of ZBTB10 promotes H3K27ac modification and transcription of TFR1
ZBTB10 is a transcriptional repressor [21]. Following treatment of M1-polarized macrophage exosomes with HK2 cells, ZBTB10 levels decreased, accompanied by reduced repressive activity. Consequently, the expression of the ferroptosis-promoting gene TFR1 is expected to increase when ZBTB10 levels are low. After confirming the interference efficiency of ZBTB10 at mRNA and protein levels (Figure 5 A), RT-qPCR results showed that TFR1 mRNA levels were significantly upregulated following ZBTB10 interference (Figure 5 B), confirming the regulatory effect of ZBTB10 on TFR1. Further, luciferase reporter assays indicated no change in TFR1 promoter activity following ZBTB10 interference (Figure 5 C). Thus, we hypothesized that TFR1 undergoes histone modification and that ZBTB10 knockdown activates H3K27ac modification of TFR1. Using ChIP, we assessed H3K27ac modification levels in the TFR1 promoter region following ZBTB10 interference (Figure 5 D). The results showed that ZBTB10 knockdown activated H3K27ac modification in the TFR1 promoter region. We designed primers every 400 bp in the promoter region, identifying five potential sites. ChIP results showed binding at all five sites, with the highest binding at site 5 (Figure 5 E).
Figure 5
Downregulation of ZBTB10 promotes h3k27ac modification and transcription of TFR1. A – RT-qPCR and Western blot analysis of ZBTB10 expression in HK2 cells after ZBTB10 interference. B – RT-qPCR analysis of TFR1 expression in HK2 cells after ZBTB10 interference. C – Luciferase reporter assay to detect the promoter activity of TFR1 in HK2 cells after ZBTB10 interference. D – ChIP assay to detect H3K27ac modification levels in the TFR1 promoter region in HK2 cells after ZBTB10 interference. E – ChIP assay to detect ZBTB10 binding at five potential H3K27ac modification sites in the TFR1 promoter region
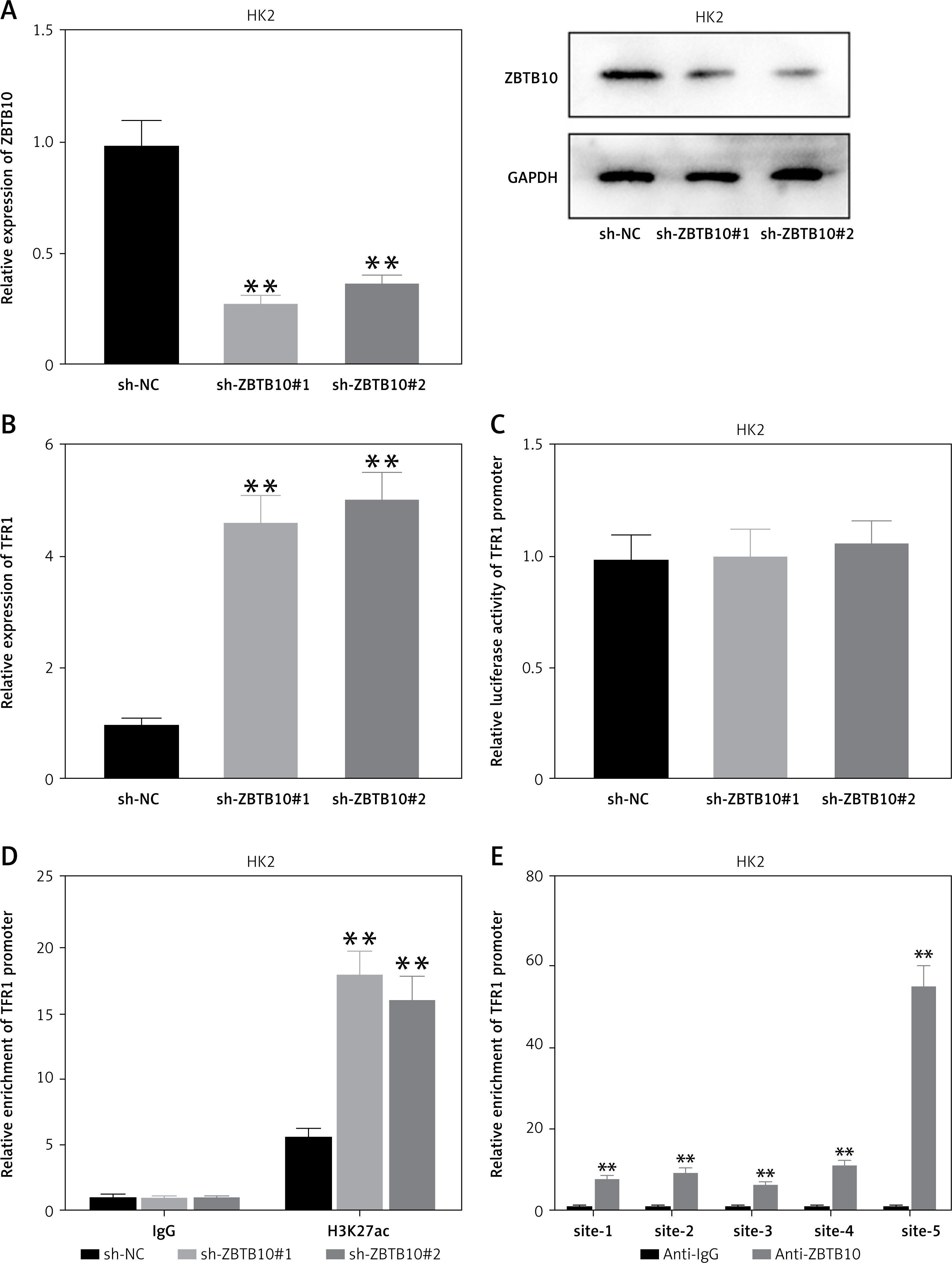
These findings collectively demonstrate that M1-polarized macrophage exosomes deliver miR-582-5p to HK2 cells, inhibiting the expression of the transcriptional repressor ZBTB10. The loss of ZBTB10 induces H3K27ac modification of the key ferroptosis gene TFR1, promoting TFR1 transcription and activating ferroptosis in HK2 cells.
Discussion
This study demonstrated that exosomes derived from M1-polarized macrophages inhibit the proliferation of HK2 cells and promote ferroptosis, offering new insights into their role in S-AKI.
Our findings align with previous research highlighting the significance of macrophage exosomes in intercellular communication and modulation of cellular functions. For instance, macrophage-derived exosomes have been shown to influence endothelial cell function and contribute to vascular inflammation [22, 23]. Similarly, our study suggests that M1 macrophage exosomes contain bioactive molecules that critically affect HK2 cell viability and oxidative stress, culminating in ferroptosis.
A pivotal discovery in our research is the role of miR-582-5p in mediating the effects of M1 macrophage exosomes on HK2 cells. MiRNAs are recognized for their regulatory roles in gene expression and involvement in various pathological processes, including inflammation and cell death [24]. Our data reveal that miR-582-5p is significantly upregulated in exosome-treated HK2 cells and is crucial in inhibiting cell proliferation and promoting ferroptosis. This observation is consistent with previous studies highlighting the significance of miRNAs in exosome-mediated cellular effects.
The selective upregulation of TFR1, a gene associated with ferroptosis, by miR-582-5p, without affecting other related genes (NRF2, GPX4, ACSL4), indicates a targeted regulatory mechanism. This specificity likely arises from the direct binding of miR-582-5p to the 3′UTR of ZBTB10, a transcriptional repressor, resulting in its downregulation. This targeted interaction is supported by other studies showing that miRNAs exert their effects through specific target interactions.
Moreover, our mechanistic studies indicate that the downregulation of ZBTB10 leads to increased TFR1 expression through enhanced H3K27ac modification at the TFR1 promoter region. Histone modifications play a pivotal role in gene regulation [25, 26], and our findings add to the growing evidence linking epigenetic changes to ferroptosis. This mechanism is in agreement with prior observations that histone modifications can influence gene expression in various cellular processes, including ferroptosis.
The implications of our study are substantial for understanding the pathogenesis of S-AKI. Ferroptosis is increasingly recognized as a critical cell death pathway in various diseases, including AKI. By elucidating the role of M1 macrophage exosomes and miR-582-5p in promoting ferroptosis, our research identified potential therapeutic targets for mitigating kidney damage in sepsis. Targeting the miR-582-5p/ZBTB10/TFR1 axis could present a novel strategy to prevent or treat ferroptosis-related kidney injury.
However, there are limitations to our study that necessitate further investigation. While our in vitro findings provide compelling evidence for the role of miR-582-5p and ZBTB10 in ferroptosis, in vivo studies are needed to validate these mechanisms in the context of S-AKI. Additionally, exploring other potential targets of miR-582-5p and the broader impact of M1 macrophage exosomes on kidney function will be crucial for a comprehensive understanding of these pathways.
In conclusion, our study identified M1-polarized macrophage-derived exosomes as key mediators of ferroptosis in renal tubular epithelial cells via the delivery of miR-582-5p. This miRNA targets ZBTB10, leading to the upregulation of TFR1 and induction of ferroptosis. These findings enhance our understanding of the molecular interactions between macrophages and renal cells in S-AKI and highlight potential therapeutic targets for preventing ferroptosis-related kidney damage. Further research is required to explore the clinical implications and therapeutic potential of targeting the miR-582-5p/ZBTB10/TFR1 axis in sepsis and related conditions.