
Introduction
Postpartum depression (PPD) is a significant emotional disorder that occurs after childbirth, commonly presenting with persistent sadness, feelings of helplessness, anxiety, and fatigue, and in some cases may involve suicidal thoughts [1, 2]. The global prevalence of PPD varies considerably, posing a significant health burden, and recent estimates indicate that rates can reach 33.5% in some cohorts [3]. This widespread psychiatric disorder profoundly undermines maternal well-being following childbirth and can also compromise child health [4].
While current research on PPD has made some progress, a comprehensive understanding of its precise biological mechanisms remains limited. Genome-wide association studies (GWAS) serve as essential tools for identifying genetic markers associated with PPD across diverse populations, thereby revealing its genetic basis [5]. The accumulation of these research findings will lead to a more accurate biological understanding of PPD, paving the way for improved preventive measures and therapeutic interventions.
Mendelian randomization (MR) leverages genetic variation as instrumental variables (IVs) to infer whether an exposure has a causal effect on an outcome [6, 7]. By integrating GWAS summary statistics with expression quantitative trait loci (eQTL) information, MR has proven to be a robust method for identifying novel druggable gene targets [8]. eQTLs mapped to loci of druggable genes are often used as proxy instruments, since gene expression serves as a stable, lifelong exposure [9]. Consequently, we performed a comprehensive MR analysis of the drug-targetable genome to uncover candidate treatment targets for PPD. At the first step, we collected information on drug-targetable genes and selected those situated in blood and brain eQTLs. These selected genes were then analyzed using two-sample MR and summary-data-based MR (SMR) methods in combination with PPD GWAS data to identify genes associated with PPD.
Target genes could heighten the likelihood of developing PPD. Previous research has shown that PPD is associated with alterations in specific brain connectivity patterns, particularly in areas involved in mood regulation and stress response [10, 11]. However, the potential mediating role of brain connectivity in the pathway from target gene expression to PPD remains unclear. Understanding this relationship may elucidate the key neurobiological pathways underlying PPD. Specifically, it raises the question of whether changes in brain connectivity act as a mediator from genetic variations to PPD. Exploring this mediating pathway can not only deepen our insight into the etiology of PPD but also provide new methods for identifying biomarkers and developing targeted therapeutic interventions.
Material and methods
The flowchart of this study is presented in Figure 1.
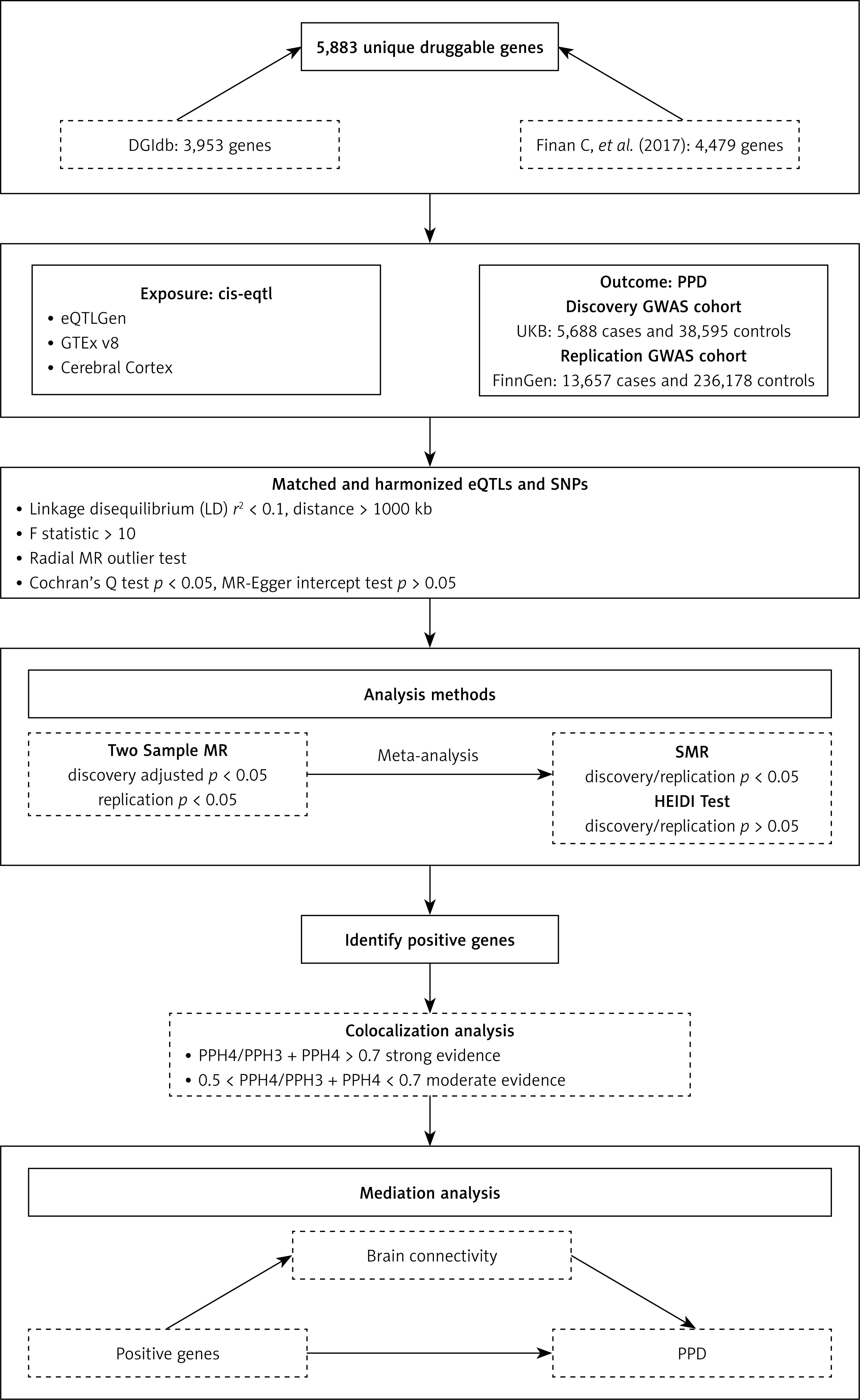
Druggable genes
We sourced the list of druggable genes from the Drug–Gene Interaction Database (DGIdb; https://dgidb.org/) by downloading its ‘Categories Data’ release (updated February 2022) [12]. We then augmented this collection with genes highlighted in the recent review by Finan et al. [13]. After merging these two lists and removing duplicates, we established a comprehensive set of druggable genes supported by previous research [14].
Exposure data
We acquired the druggable genes data from three distinct sources: blood eQTLs from eQTLGen, eQTLs from 13 brain-related tissues from the Genotype-Tissue Expression (GTEx) v8, and cerebral cortex eQTLs. The eQTLGen dataset, obtained from the eQTLGen Consortium (https://eqtlgen.org/), includes 16,987 cis-eQTLs derived from 31,684 blood samples, primarily from healthy individuals of European descent. This dataset provides detailed allele frequency statistics and comprehensive gene-level cis-eQTL data [15]. Subsequently, we assessed brain-specific gene expression using eQTL data acquired from the GTEx website (https://gtexportal.org/home/) [16], which includes expression profiles across 838 donors, encompassing 17,382 samples from across 52 different tissues and two cell lines. We focused specifically on brain-related eQTLs to investigate genes potentially linked to PPD risk. We utilized eQTL data from 13 brain-related tissues available in the GTEx v8 dataset (specific tissues listed in Supplementary S1). The cerebral cortex dataset was derived from Qi et al.’s meta-research, which used RNA sequencing data from 2,865 cerebral cortex specimens obtained from 2,443 unrelated donors of European ancestry. This study incorporated comprehensive genome-wide SNP information, providing a robust resource for exploring eQTL mapping in brain tissue (https://yanglab.westlake.edu.cn/data/brainmeta/cis_eqtl/) [17]. Detailed information is provided in Table I.
Table I
Details of eQTL and GWAS used in the study
| Dataset | Sample size | Ancestry | Consortium | Web link |
|---|---|---|---|---|
| Blood eQTL | 31684 | European | eQTLGen Consortium | https://eqtlgen.org/ |
| Brain eQTL | 17,382 | European | GTEx Consortium | https://gtexportal.org/home/ |
| Cerebral cortex eQTL | 2,865 | European | Qi et al. | https://yanglab.westlake.edu.cn/data/brainmeta/cis_eqtl/ |
| PPD GWAS (UKB) | 44283 | European | UK Biobank | https://www.nealelab.is/uk-biobank |
| PPD GWAS (FinnGen) | 249835 | European | FinnGen | https://www.finngen.fi/en |
| Smoking status GWAS | 454429 | European | Jiang L et al. | https://www.ebi.ac.uk/gwas/studies/GCST90042704 |
| Alcohol drinking GWAS | 64,001 | European | Jiang L et al. | https://www.ebi.ac.uk/gwas/studies/GCST90041731 |
| Body mass index GWAS | 461,460 | European | MRC-IEU | https://gwas.mrcieu.ac.uk/datasets/ukb-b-19953/ |
| Time spent doing moderate physical activity GWAS | 64,949 | European | MRC-IEU | https://gwas.mrcieu.ac.uk/datasets/ukb-b-2115/ |
Outcome data
GWAS data for PPD in the discovery phase were obtained from the UK Biobank, which included 5,688 patients and 38,595 controls from the European population [18]. GWAS data for the PPD replication cohort, comprising 13,657 cases and 236,178 controls, were obtained from FinnGen Consortium (https://www.finnngen.fi/en) [19]. PPD was defined as depression possibly related to childbirth. The criteria for diagnosing PPD include having given birth and meeting International Classification of Diseases, Tenth Edition (ICD-10) code F32, F33, or F53.0. This MR study leveraged publicly available GWAS summary data, relying solely on aggregated results without individual-level information, and thus did not require additional ethical approval. Specific data information and download links are shown in Table I.
Mendelian randomization analysis
The MR analysis is based on three core assumptions: (1) The instrumental variables (IVs) must be strongly associated with the exposure; (2) IVs must not have associations with potential confounders; and (3) IVs should affect the outcome exclusively via their influence on the exposure, without having any direct impact on the outcome itself [20]. In this MR study, we implemented several quality control procedures in order to ensure the reliability of the results. First, both the exposure and outcome data were derived from participants of European descent, thus reducing biases related to population stratification [21]. To ensure that druggable genes are strongly associated with the SNP, the false discovery rate (FDR) < 0.05 was chosen as the significance threshold. In addition, SNPs exhibiting linkage disequilibrium (LD) were removed from the analysis. The LD must meet the criterion of r2 < 0.1 and distance > 1,000kb. Moreover, to mitigate bias from weak instrumental variables, those with an F statistic below 10 (F = beta2/SE2) were excluded from the analysis [22]. Furthermore, the radial MR method was employed to exclude genes in which the explained variance in the PPD exceeded the variation explained by the exposure [23].
We conducted MR analysis using the R package TwoSampleMR (version 0.5.7). The exposure and outcome datasets were loaded and harmonized via the package’s harmonise_data function. The inverse variance weighted (IVW) method was adopted as the principal analytical approach, and the Wald ratio test was used when only a single SNP instrument was available. To address multiple testing and reduce the risk of false-positive results, we applied the FDR adjustment to the p-values. We also performed meta-analysis on the IVW results for the discovery and replication cohorts using the meta package in R, selecting either a random- or fixed-effects model based on the I2 value and the heterogeneity p-value [24]. To verify the robustness of the results, we performed sensitivity analysis with the MR Egger, weighted median, simple mode, and weighted mode models. Additionally, we assessed heterogeneity and directional pleiotropy between IVs using Cochran’s Q test and MR-Egger intercept test, with p > 0.05 indicating not susceptible to bias in heterogeneity and directional pleiotropy.
Summary-data-based MR (SMR)
SMR analysis was performed to validate the causal relationships between specific genes and PPD by integrating eQTL-based gene expression data. By focusing on the most strongly associated cis-QTLs, SMR achieves markedly greater power than conventional MR analyses, especially when dealing with independent datasets that have large sample sizes [25]. This methodological approach enhances the reliability and stability of the results. Furthermore, the heterogeneity in the dependent instruments (HEIDI) test, which utilizes multiple SNPs within a designated region, is designed to differentiate between a true causal association of genes with PPD risk and associations driven by linkage disequilibrium or horizontal pleiotropy [25]. A p-value < 0.05 was considered significant for SMR analysis, while a HEIDI p-value > 0.05 suggested that the gene-PPD correlation was not driven by LD. Both SMR and HEIDI analyses were conducted using the SMR software tool, version 1.3.1.
Colocalization analysis
We conducted colocalization analyses with the coloc R package to identify shared causal variants between PPD and previously identified eQTLs. The analysis involves assessing five posterior probabilities, with each probability linked to a particular hypothesis: 1) H0: No genetic variants influence either of the two traits; 2) H1: Only gene expression is affected by a specific genetic variant; 3) H2: Only disease risk is influenced by a particular genetic variant; 4) H3: Each trait is determined by distinct genetic variants; 5) H4: Both traits are jointly influenced by a common genetic variant. For the colocalization of eQTL-GWAS, a 100 kb window was applied. Prior probabilities were specified as 1 × 10–4 for causal variants linked to trait 1 (eQTL) or to trait 2 (PPD), and as 1 × 10–5 for variants affecting both traits [26, 27]. A posterior probability calculated as PPH4/(PPH4 + PPH3) greater than 0.7 was interpreted as strong evidence of colocalization, whereas a value greater than 0.5 but less than or equal to 0.7 was considered moderate evidence.
Negative control outcome analysis
To assess possible bias from population stratification and residual confounding, we undertook a negative control outcome analysis as an additional sensitivity test [28]. In this analysis, we compared the association between druggable genes and PPD with that between druggable genes and negative control outcomes. Theoretically, these negative control outcomes are not directly regulated by druggable genes, although they may be influenced by similar confounders [29]. For this analysis, we selected smoking status, alcohol consumption, body mass index, and time spent doing moderate physical activity as negative control outcomes. Although these measures have been previously linked to PPD [30–33], they should not be directly affected by druggable genes. Detailed information and download links for each negative-control trait are provided in Table I. We employed the same MR methods – including the IVW approach and other sensitivity tests – for both PPD and the negative control outcomes to confirm the reliability of our results.
Protein–protein interaction network construction
To gain deeper insights into intracellular protein–protein interactions, we built a protein–protein interaction (PPI) network employing the STRING database (https://string-db.org/). We filtered the interactions in the network using a confidence threshold of 0.4 while keeping all other parameters unchanged. This approach allowed for a systematic analysis of potential functional associations among proteins [34, 35].
Mediation analysis
Studies have shown an association between brain connectivity and PPD, so we applied MR analysis to assess the potential causal impact of brain connectivity on PPD. SNPs associated with brain connectivity were selected using a threshold of p < 5 × 10–6, and the criterion for LD was r2 < 0.001 with a distance > 1,000 kb. Furthermore, we propose that druggable genes may affect PPD by affecting brain connectivity. To evaluate this possibility, we performed mediation MR analyses to estimate the proportion of the effect of the druggable genes on PPD that is mediated via alterations in brain connectivity.
Using a two-step mediated MR approach, we calculated the mediation effect using coefficient multiplication, and defined the mediation proportion as the mediated effect divided by the overall effect. The standard error (SE) and 95% confidence interval (CI) for the mediated effect were calculated using the delta method, which accounts for measurement errors to improve the reliability of the results. By distinguishing between the overall effects of eQTLs on PPD and the specific mediating effects involving brain connectivity, this analysis provides deeper insights into the underlying causal pathways.
Results
Druggable genes
By combining data from the DGIdb [12] and a prior literature review [13], we identified 5,883 unique druggable genes, as designated by the Human Genome Organisation Gene Nomenclature Committee, for subsequent analyses (Supplementary Table SII).
Mendelian randomization analysis
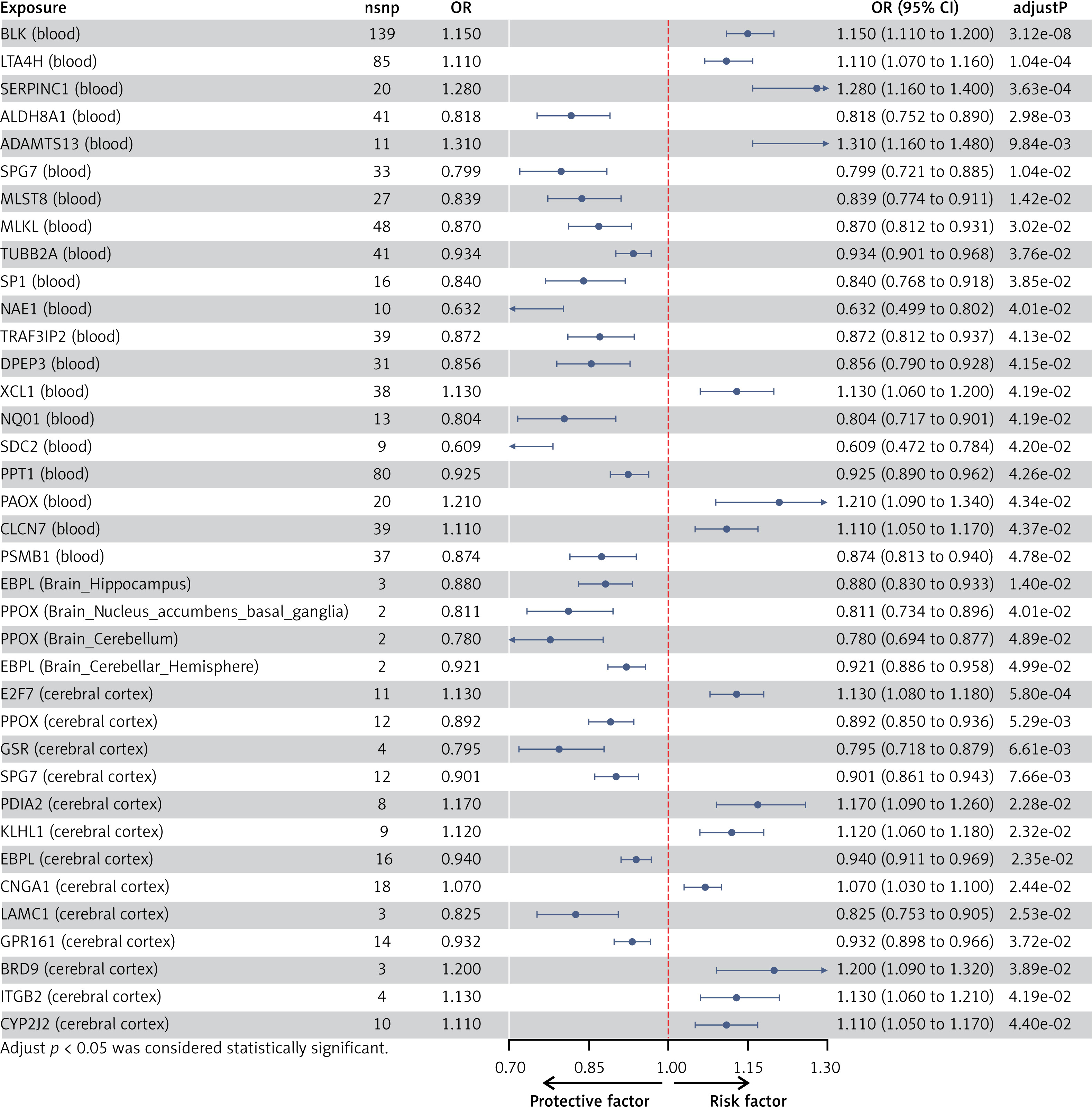
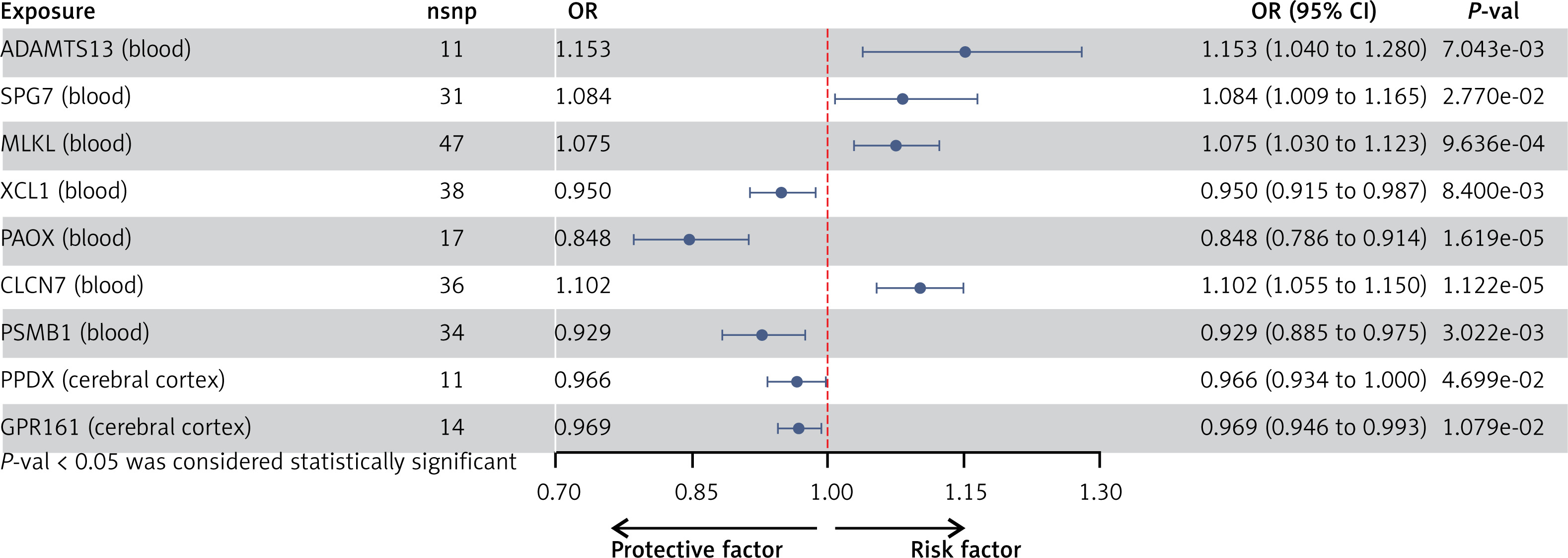
After implementing several quality control procedures, we identified 2,694 gene symbols from blood eQTLs, 2,325 from 13 brain-related tissues from the GTEx, and 2,156 from the cerebral cortex. MR analysis identified 37 significant genes associated with PPD, including 20 from blood eQTLs, four from the GTEx (Brain_Hippocampus, Brain_Nucleus_accumbens_basal_ganglia, Brain_Cerebellum, and Brain_Cerebellar_Hemisphere tissues), and 13 from cerebral cortex (Figure 2). Detailed information for the significant IVs and full MR results can be found in Supplementary Tables SIII–SV. As shown in Figure 2, the expression of these genes was causally associated with PPD risk (FDR < 0.05). In the replication phase, we confirmed the causal association between the genetically predicted expression of 9 genes and PPD risk (p < 0.05), as shown in Figure 3. Among them, seven genes – ADAMTS13, SPG7, MLKL, XCL1, PAOX, CLCN7, and PSMB1 – showed significance in blood tissue (ADAMTS13: OR = 1.153, SPG7: OR = 1.084, MLKL: OR = 1.075, XCL1: OR = 0.950), while two genes, PPOX and GPR161, demonstrated significance in the cerebral cortex tissue (PPOX: OR = 0.966 and GPR161: OR = 0.969) based on the replication cohort. Additionally, the meta-analysis of the IVW results from both the discovery and replication cohorts revealed that four genes remained positive. Among them, three genes – ADAMTS13 (OR = 1.156, 95% CI: 1.068–1.251, p < 0.001), CLCN7 (OR = 1.104, 95% CI: 1.068–1.142, p < 0.001), PSMB1(OR = 0.912, 95% CI: 0.876–0.949, p < 0.001) – showed significance in blood tissue, while GPR161 (OR = 0.957, 95% CI: 0.938–0.977, p < 0.001) was significant in cerebral cortex tissue (Table II). Further details of the MR results can be found in Supplementary Tables SVI–SIX. All identified genes also passed sensitivity, Cochran’s Q, and MR-Egger intercept tests.
Table II
Meta-analysis of IVW results from discovery and replication cohorts
Summary-data-based MR (SMR)
We further confirmed the association related to the gene expression level and PPD risk of the positive genes identified in the above process using SMR and HEIDI tests. Upon comprehensive analysis, out of the nine genes that were positively identified in both the primary and replication cohorts, only one gene – CLCN7, from blood – maintained a significant link with PPD risk. Specifically, CLCN7 showed an OR of 1.102 (P-SMR = 0.0189, P-HEIDI = 0.585), based on the results from the discovery cohort SMR analysis. Based on these results, CLCN7 passed the test and was selected as a robust candidate for further investigation. Detailed results are presented in Table III and Supplementary Tables SX–SXV.
Colocalization analysis
Colocalization analysis was employed to evaluate whether associations for the exposure and the outcome arise from the same causal SNP. Previous evidence suggests that eQTLs identified through both MR and colocalization analyses are more likely to serve as drug targets, with a higher likelihood of successful approval [8]. Our results indicated that CLCN7 demonstrated a posterior probability of 74.9% based on the discovery cohort, providing strong evidence of colocalization (Table III).
Negative control outcome analysis
For the negative control outcome analysis, we performed MR analysis to assess the associations between the CLCN7 and four negative outcomes – smoking status, alcohol consumption, body mass index, and time spent engaging in moderate physical activity. The IVW analyses produced p-values above 0.05 for every trait, indicating that none of these associations reached statistical significance. Detailed results are provided in Supplementary Table SXVI.
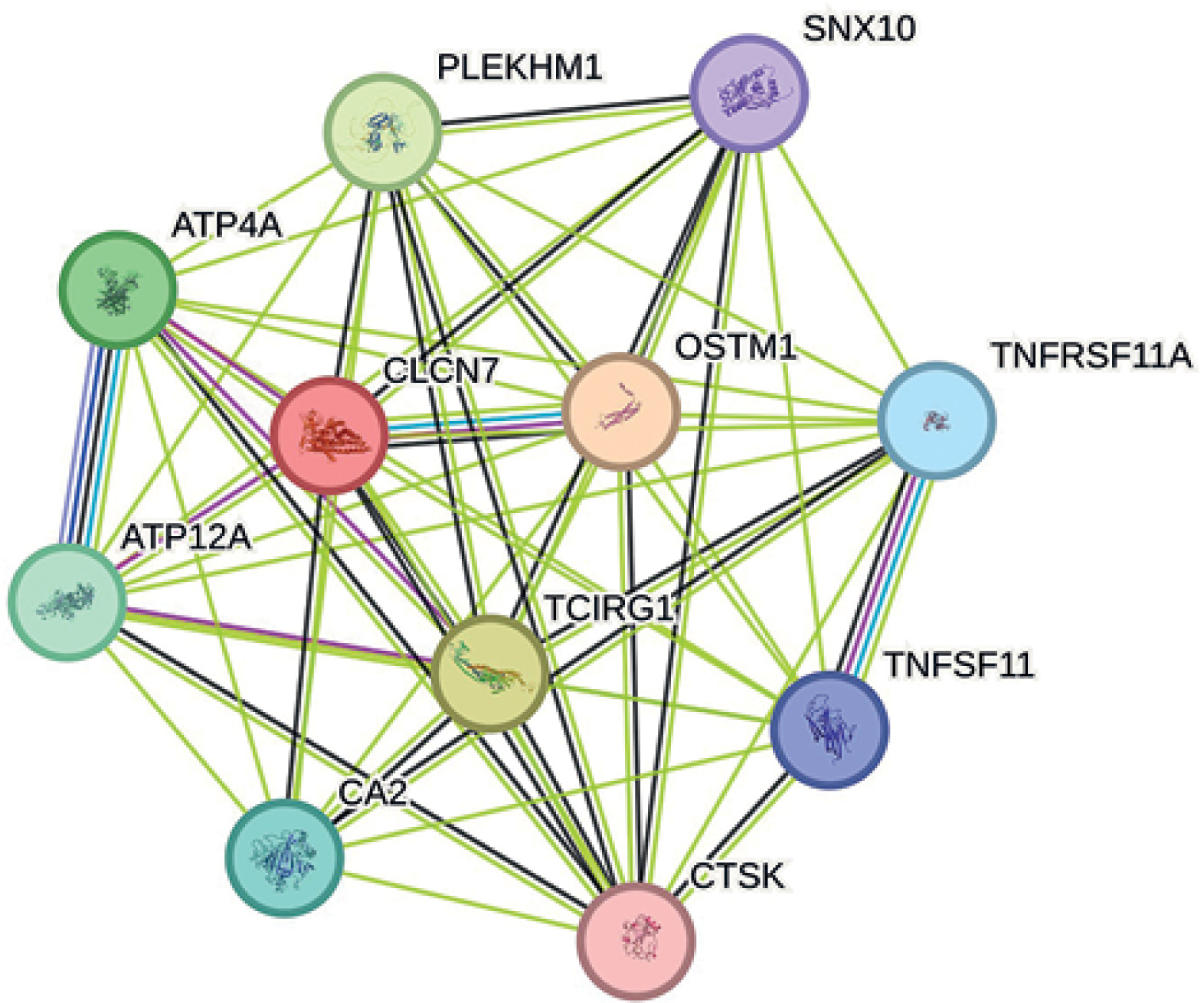
PPI networks
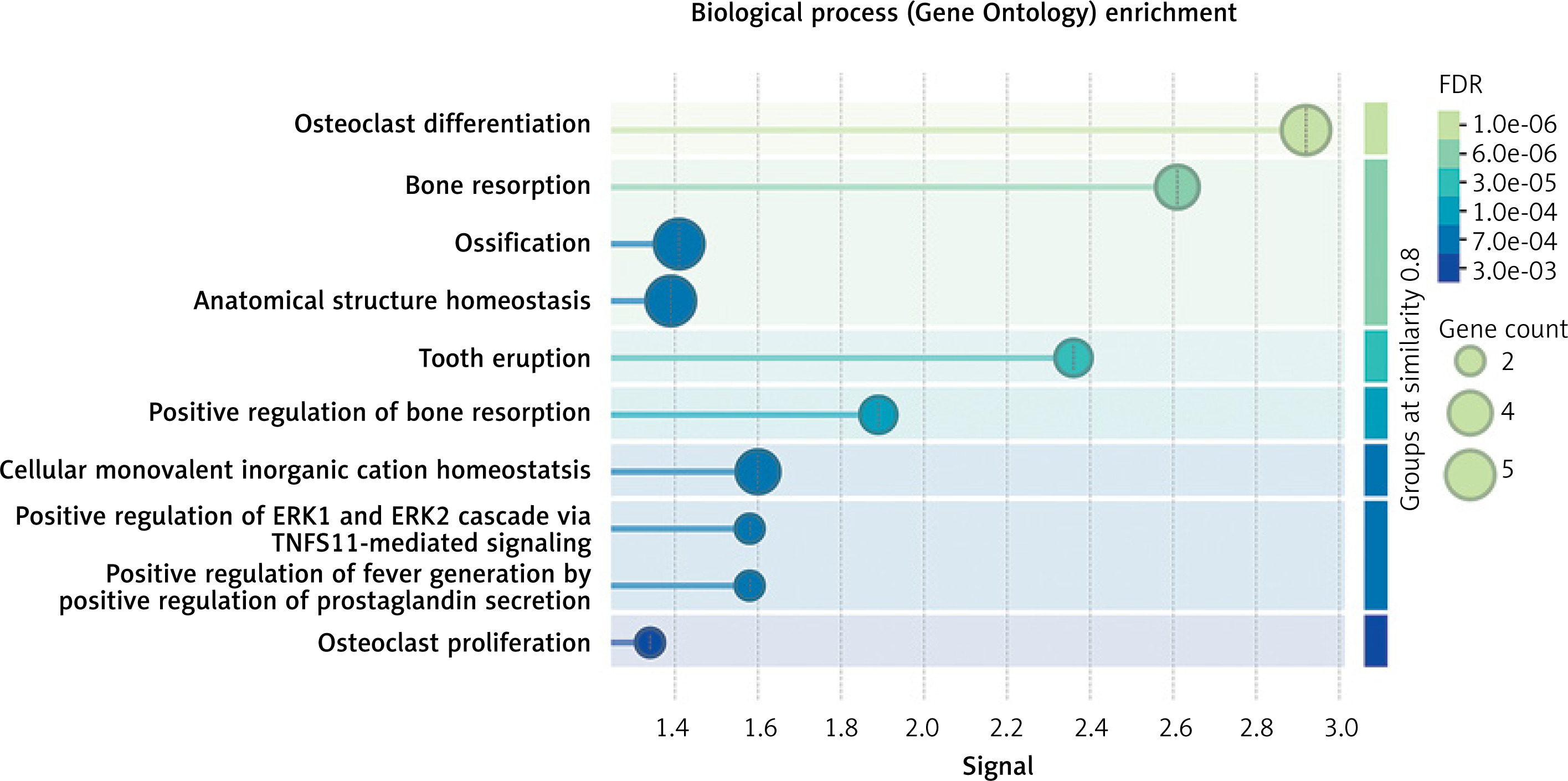
The drug target gene CLCN7 was imported into the STRING (https://cn.string-db.org/) database to construct a network. Figure 4 shows that CLCN7 is connected to the gene products of OSTM1, TCIRG1, SNX10, PLEKHM1, TNFRSF11A, TNFSF11, and others. Functional analysis of this network delineates the roles and functions of the drug target and its associated genes, indicating that the network is involved in osteoclast differentiation, lysosomal acidification, intracellular homeostasis and positive regulation of the ERK1 and ERK2 cascade via TNFSF11-mediated signaling (Figure 5).
Mediation analysis
MR mediation analyses were conducted to assess whether brain connectivity mediates the relationship between the identified positive genes and PPD. The results indicated that five brain connectivity measures had significant causal effects on PPD. These five connectivity measures were incorporated into the mediation analysis for the identified key gene, CLCN7, in relation to PPD (Supplementary Table SXVII).
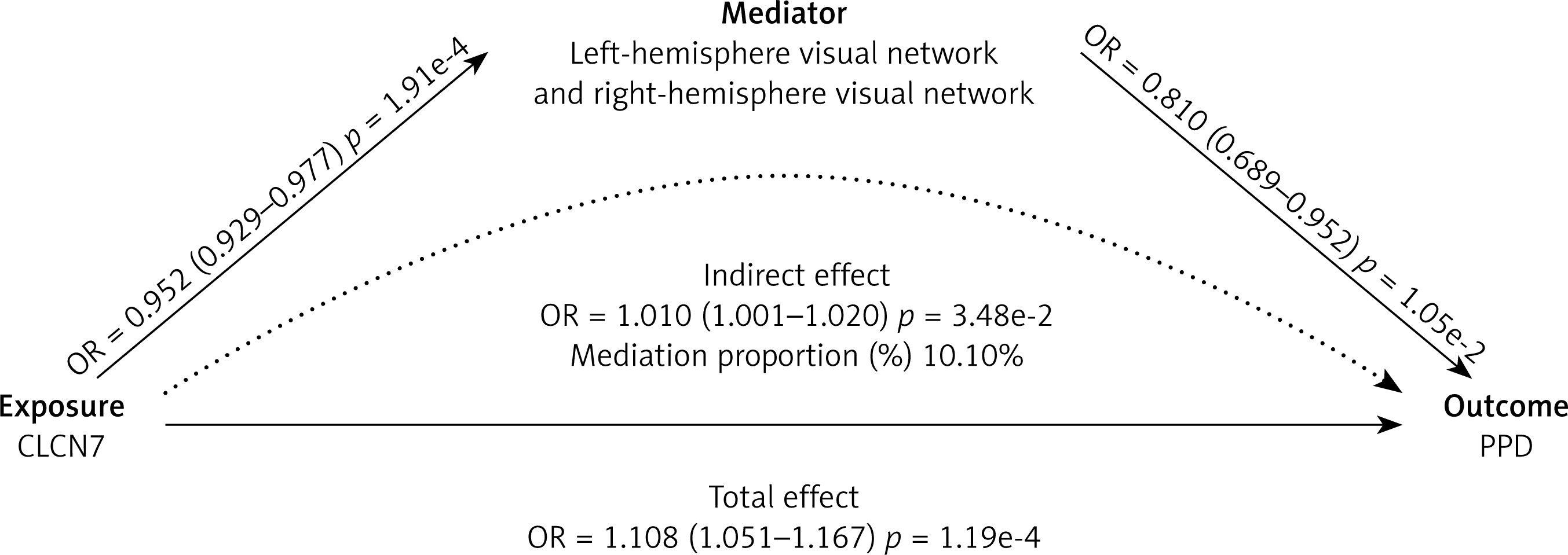
Among these, CLCN7 was found to exhibit causal relationships with left- to right-hemisphere visual network white-matter structural connectivity (β = –0.049, 95% CI: –0.074 to –0.023, p < 0.001), as well as the left-hemisphere visual network to right-hemisphere dorsal attention network white-matter structural connectivity (β = –0.024, 95% CI: –0.046 to –0.002, p = 0.028) (Figure 6, Supplementary Table SXVIII). Based on these findings, the white-matter structural connectivity between the left- and right-hemisphere visual networks (β = 0.103, 95% CI: 7.33e-4 to 0.019, p =0.035) partially mediates the effect of CLCN7 on increased PPD, with a mediation proportion of 10.10%. However, the connectivity between the left-hemisphere visual network and right-hemisphere dorsal attention network (β = 5.69e-3, 95% CI: –1.86e-3 to 0.013, p = 0.140) does not show a significant mediation effect (Supplementary Table SXIX).
Discussion
In this study, we combined large-scale genetic datasets and employed MR, SMR, the HEIDI test and colocalization analyses to identify and confirm druggable genes potentially contributing to PPD risk. Adopting a multi-step approach including discovery, replication, and functional validation, we sought to ensure more reliable causal inferences. Ultimately, we identified one gene, CLCN7, as a likely causal factor for PPD and demonstrated that the left- to right-hemisphere visual network white-matter structural connectivity mediates a portion of the genetic risk conferred by CLCN7.
Based on these genetic findings, we explored the potential biological role of CLCN7 in the underlying mechanisms of mood disorders. CLCN7 is considered a biologically plausible candidate in relation to mood regulation and the neurobiological pathways involved in depression. CLCN7 encodes a chloride channel critical for lysosomal acidification and maintaining cellular homeostasis [36]. Impairments in lysosomal functions are increasingly associated with psychiatric disorders, possibly impacting neurotransmitter turnover, neuronal plasticity, and the preservation of synaptic integrity [36]. Studies suggest that CLCN7-related lysosomal dysfunction can hinder neuronal survival, disrupt synaptic processes, and contribute to both neurodegenerative and psychiatric disorders [37]. Although the precise mechanisms connecting CLCN7 to PPD remain unclear, our results may pave the way for future functional studies. Employing animal models and single-cell RNA sequencing could elucidate how CLCN7 dysregulation affects neural circuits and cell-specific pathways, potentially leading to targeted therapies for PPD.
Notably, CLCN7 demonstrated a potential influence on the risk of postpartum depression (PPD) by altering brain connectivity patterns. Specifically, white matter structural connectivity between left- and right-hemisphere visual networks partially mediated the genetic risk associated with CLCN7. Our findings indicate that enhanced left- to right-hemisphere visual network white-matter structural connectivity generally offers protection against PPD; however, higher expression levels of the CLCN7 gene can diminish this connectivity, thereby elevating the risk of developing PPD. Additional neuroimaging investigations using techniques such as functional magnetic resonance imaging (fMRI) are required to further characterize these circuits and validate our results.
Recent research suggests that disruptions in sensory integration and the regulation of attention may affect emotional processing and responses to stress [11, 38]. By determining that part of the genetic predisposition operates through these connectivity patterns, we present initial evidence for a neurobiological foundation that links genetic variations to PPD. This discovery also highlights the complexity of PPD, indicating that it is not solely associated with traditional mood-regulatory circuits (such as the limbic system and prefrontal cortex) but may also be influenced by more extensive, integrative neural networks [39, 40].
Additionally, negative control outcome analyses supported the reliability of these associations by showing no significant link between CLCN7 and unrelated outcomes (e.g., smoking status, alcohol consumption, BMI, and time spent engaging in moderate physical activity), thus reducing concerns about residual confounding or population stratification. Meanwhile, PPI analysis revealed that CLCN7 interacts with multiple proteins (e.g., OSTM1, TCIRG1, SNX10, PLEKHM1, TNFRSF11A, and TNFSF11), indicating its roles in lysosomal acidification, intracellular homeostasis, and the positive regulation of the ERK1 and ERK2 cascade via TNFSF11-mediated signaling, thus suggesting a potential involvement in the etiology of depression [37, 41]. Overall, these results further reinforce the causal role of CLCN7 in postpartum depression and highlight valuable targets for potential therapeutic intervention.
Another important aspect of this work is its potential translational relevance. After starting with a collection of druggable genes, we employed MR, SMR, and colocalization analysis, and identified high-confidence candidate genes that may reduce the risk of PPD. While immediate clinical applications remain distant, the identification of druggable targets opens the door to future pharmacological interventions. Targeting CLCN7-related pathways might prove beneficial in preventing or treating PPD, especially for patients at heightened genetic risk. Future research should prioritize the functional validation of CLCN7 to evaluate its therapeutic potential, constituting a critical step toward developing targeted treatments for PPD.
Despite these strengths, a number of limitations should be acknowledged. First, our analyses primarily rely on GWAS and eQTL datasets derived from individuals of European ancestry, potentially limiting generalizability to other populations. Second, although rigorous methods (such as HEIDI and colocalization tests) were employed, residual confounding, selection bias, and horizontal pleiotropy cannot be completely excluded. Third, environmental variables – such as stress and social support, known to critically influence PPD – were not included in our analyses. Future research should integrate these variables to evaluate their modulation of genetic risks associated with CLCN7 and other relevant genes. Lastly, while we identified brain connectivity patterns as mediators, the specific molecular and cellular mechanisms remain unclear; further experimental research using techniques such as single-cell transcriptomics, imaging genetics, and animal models is needed to clarify these mechanisms.
In conclusion, this study provides evidence that CLCN7 plays a potential causal role in the genetic architecture of PPD. The partial mediation of CLCN7’s effect on PPD through left- to right-hemisphere visual network white-matter structural connectivity patterns emphasizes the importance of integrating genetic and neuroimaging data to understand the pathogenesis of complex psychiatric conditions. Our findings pave the way for future mechanistic research and offer the possibility of developing targeted interventions to reduce the burden of PPD.