Introduction
Epilepsy ranks as the second most common neurological disorder worldwide, affecting over 65 million patients globally [1]. Temporal lobe epilepsy (TLE) is the most common type of epilepsy [2, 3]. The incidence of TLE is 32 to 82 cases per 10,000. Status epilepticus induces damage of the nervous system and hippocampal neurons [4]. The progression of epilepsy promotes the increase in lipid peroxidation, ischaemia, inflammation, and oedema, which are the key factors for the loss of hippocampal neurons, and hippocampal sclerosis [1, 5]. Although great advances have been made in the treatment of epilepsy, such as anticonvulsant drugs (AEDs), long-term use of AEDs may induce drug-resistant epilepsy [6, 7]. Therefore, developing a novel strategy for epilepsy is urgently needed.
Ketogenic diets (KD) (high-fat, adequate-protein, and low-carbohydrate diets) are a non-pharmacological treatment designed to mimic the effects of starvation in drug-resistant individuals [8]. The benefits of a KD in paediatric patients include improved seizure control, weight loss, and improved insulin sensitivity [9]. Ketones may confer neurologic protection. Ketone bodies exert anti-oxidant and anti-inflammatory effects, as well as inducing cellular, epigenetic, and gut-microbiome alterations. KD show a positive impact on behavioural and cognitive functions [10]. However, the underlying mechanisms are still unknown.
Ferroptosis is a form of programmed cell death, characterized by lipid peroxidation and iron overload [11]. Ferroptosis is regulated by three pathways: oxidative stress, lipid peroxidation, and iron metabolism [12, 13]. The activation of ferroptosis-related pathways is collectively involved in the pathogenesis of epilepsy. For instance, the hypoxia inducible factor-1α (HIF-1α)/haem oxygenase-1 (HO-1) pathway suppresses neuronal ferroptosis in epilepsy via inhibiting oxidant stress [14]. Neurotoxic A1-mediated lipid peroxidation promotes the ferroptosis of neurons in epilepsy via downregulating solute carrier family 7a member 11 (SLC7A11) [15]. Mitochondrial ferritin, which functions as an iron transporter, suppresses ferroptosis of neurons through maintaining iron homeostasis [16].
This study aimed to investigate the effects of KD on the neuron functions in the progression of epilepsy. The effects of KD on iron homeostasis and neuron functions may shed new light on the progression of epilepsy.
Material and methods
Cell culture
Mouse hippocampal neuronal cells (HT22) were provided by Procell (Wuhan, China). Cells were incubated at 37°C in an incubator with 5% CO2 using Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), and low-glucose DMEM was used for comparison.
Cells were treated with Mg2+, erastin, RSL3, and Fer-1, as well as the inhibitor of apoptosis (z-VAD-fmk), pyroptosis (PBzyme) and necroptosis (Nec-1s).
RT-qPCR
Cells were lysed using TRIzol buffer (R0016; Beyotime, China) and the total RNA was collected. RNA concentration and purity were detected using a UV spectrophotometer (Bio-Rad, USA). Then RNA was reverse transcribed into cDNA using a reverse transcription kit (RR037B; Takara, Japan). Relative mRNA expression was determined using a SYBR Premix Ex Taq kit (DRR041A; Takara, Japan) and calculated using the 2–ΔΔCq method. The sequences of the primers used in PCR were as follows: histone deacetylase 4 (HDAC4), F: 5′-CTGCAAGTGGCCCCTACAG-3′ and R: 5′-CTGCTCATGTTGACGCTGGA-3′; and glyceraldehyde-3-phosphate dehydrogenase, F: 5′-AGGTCGGTGTGAACGGATTTG-3′ and R: 5′-GGGGTCGTTGATGGCAACA-3′.
Western blot analysis
Proteins were extracted from HT22 cells. Following concentration and denaturation, the proteins were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes, which were blocked with 5% skimmed milk. Then membranes were incubated with primary antibodies, such as anti-HDAC4 (ab240643; 1 : 1000, Abcam, UK), anti-acetyl H3K9 (ab190479; 1 : 1000, Abcam, UK), and anti-transferrin receptor (TFRC) (ab214039; 1 : 1000, Abcam, UK), followed by secondary antibody (ab205718; 1 : 10000, Abcam, UK). Finally, the immunoreactive bands were captured using an enhanced chemiluminescence reagent. β-actin (ab227387; 1 : 10000, Abcam, UK) served as a loading control.
Co-immunoprecipitation (Co-IP) assay
Cells were lysed with radio-immunoprecipitation assay lysis buffer (20-188; Millipore, USA). Afterwards, the protein lysates were collected and incubated with Protein A/G magnetic beads containing antibodies against HDAC4 (ab240643; 1 : 30, Abcam, UK) and TFRC (ab214039; 1: 30, Abcam, UK). Subsequently, the beads were boiled and subjected to western blot assay.
Determination of malondialdehyde (MDA)
The MDA levels were detected in cell lysates and measured using an MDA Kit (MAK568; Sigma-Aldrich, USA). Briefly, cells were lysed and centrifuged. Then the lysates were supplemented with thiobarbituric acid. Finally, MDA levels were determined at a wavelength of 532 nm.
Determination of intracellular iron levels
The intracellular iron levels were determined using a Colorimetric Iron Assay Kit (ab83366; Abcam, UK).
Glutathione (GSH) assay
GSH levels were determined using a GSH Assay Kit (ab112132; Abcam, UK). Briefly, cells were plated in a 24-well plate. After supplementation with magnesium ion (Mg2+)-free solution, low-glucose medium and erastin, cells were lysed with GSH assay buffer. GSH was determined using a kinetic assay and calculated at a wavelength of 412 nm.
Cell Counting Kit-8 (CCK-8) assay
After transfection, cells were collected and plated in a 24-well plate. After being cultured for 48 h, the cells were treated with CCK-8 solution (96992-100TESTS-F; Sigma-Aldrich, Germany) and mixed for another 90 min at 37°C. Subsequently, cell viability was quantified by measuring absorbance at 450 nm using a microplate reader.
Propidium iodide (PI) staining
After fixation in 4% paraformaldehyde and permeabilization with 0.2% Triton X-100, cells were blocked with 5% bovine serum. Then cells were stained with PI. The image was taken using a microscope (M205 FA; Leica, Germany).
Animal experiment
Male Sprague Dawley (SD) rats (80–100 g) were purchased from Changsheng Biology (Liaoning, China). Rats had free access to food and water. This study was approved by the Animal Care Board of The First Affiliated Hospital of Harbin Medical University.
Rats were randomly divided into three groups: the sham group, the kainite acid (KA) group, and the KA + KD group. Rats in the KA group were hypodermically injected with 10 mg/kg KA. Rats in the KA + KD group were hypodermically injected with 10 mg/kg KA and administered KD formula (76% fat, 16% protein, 3% carbohydrate, and 5% dietary fibre in kcal) (Kuibuqianli Biology, Xuancheng, China). Rats in the sham group were hypodermically injected with saline.
Immunofluorescence assay
After anaesthesia, the rats were sacrificed and the brain tissues were collected. The slides were fixed in paraffin, deparaffinized and immersed in ethylenediaminetetra-acetic acid buffer. After blocking with 1% bovine serum albumin regents, the sections were incubated with primary antibodies against NeuN (ab177487; 1 : 100, Abcam, UK) and HDAC4 (ab313474; 1 : 200, Abcam, UK), and then with secondary antibody (ab150077; 1 : 200, Abcam, UK). Nuclei was counterstained with 4’,6-diamidino-2-phenylindole. Finally, the images were captured using a microscope (M205 FA; Leica, Germany).
Results
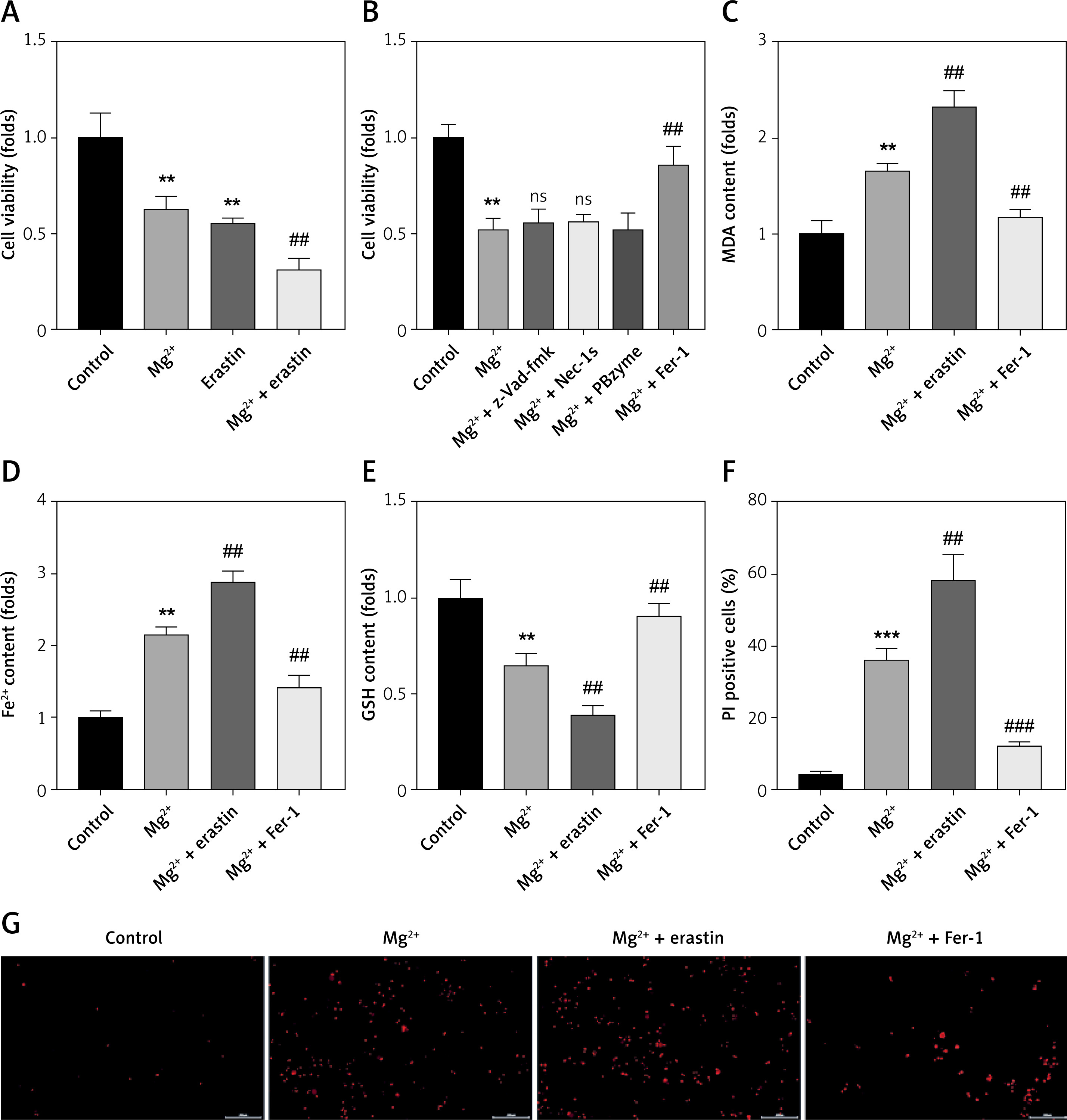
Epilepsy-induced ferroptosis-related neuronal loss
Ferroptosis-mediated neuron loss is involved in the progression of epilepsy. To further confirm this, neurons were treated with a ferroptosis inducer (erastin) as well as an inhibitor (Fer-1) after exposure to Mg2+-free solution. We found that erastin treatment significantly enhanced the effects of Mg2+ treatment and suppressed cell viability of neurons, suggesting that the progression of epilepsy may induce neuronal ferroptosis. However, epilepsy can induce other forms of cell death, such as apoptosis, pyroptosis and necroptosis. To further confirm this, cells were treated with inhibitors of apoptosis (z-VAD-fmk), pyroptosis (PBzyme) and necroptosis (Nec-1s). As shown in Figure 1 B, the decrease in the cell viability induced by Mg2+ treatment was significantly alleviated by Fer-1, which showed no significant alteration by the inhibitors of apoptosis, pyroptosis, and necroptosis. Mg2+ treatment significantly increased the release of MDA and ferrous iron (Fe2+), and decreased GSH (Figures 1 C–E), which was significantly enhanced by erastin, but reversed by Fer-1. Moreover, the increase in the percentages of PI positive cells was significantly enhanced by erastin (Figures 1 F, G), but reversed by Fer-1. These findings confirmed that the progression of epilepsy is accompanied by the ferroptosis of neurons.
Figure 1
Epilepsy-induced ferroptosis-related neuronal loss. A, B – Cell viability was detected using CCK-8 assay. C – Release of MDA was determined using MDA assay. D – Release of ferrous iron was detected using ELISA assay. E – Release of GSH was detected using GSH assay. F, G – Death of neurons was determined using PI staining
MDA – malondialdehyde (MDA), GSH – glutathione, CCK-8 – Cell Counting Kit-8, PI – propidium iodide. **P < 0.01, ***p < 0.001, ##p < 0.01, ###p < 0.001.
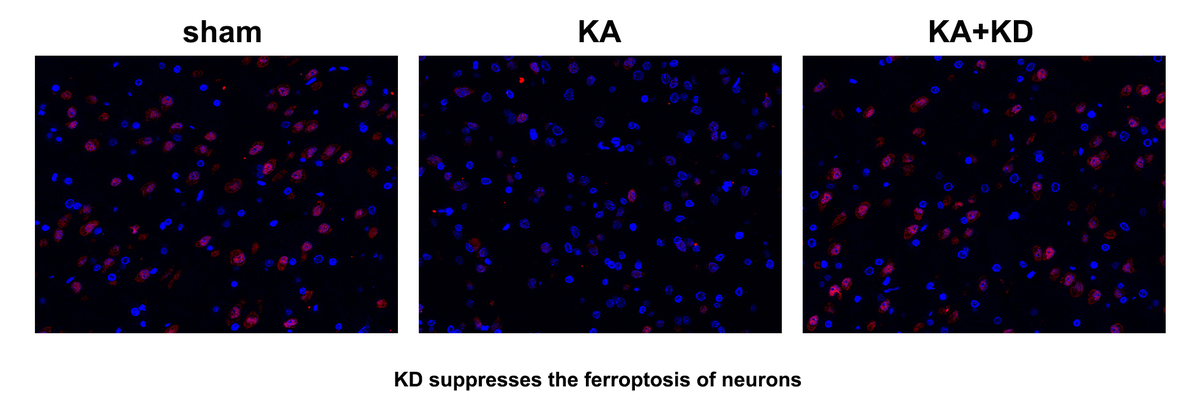
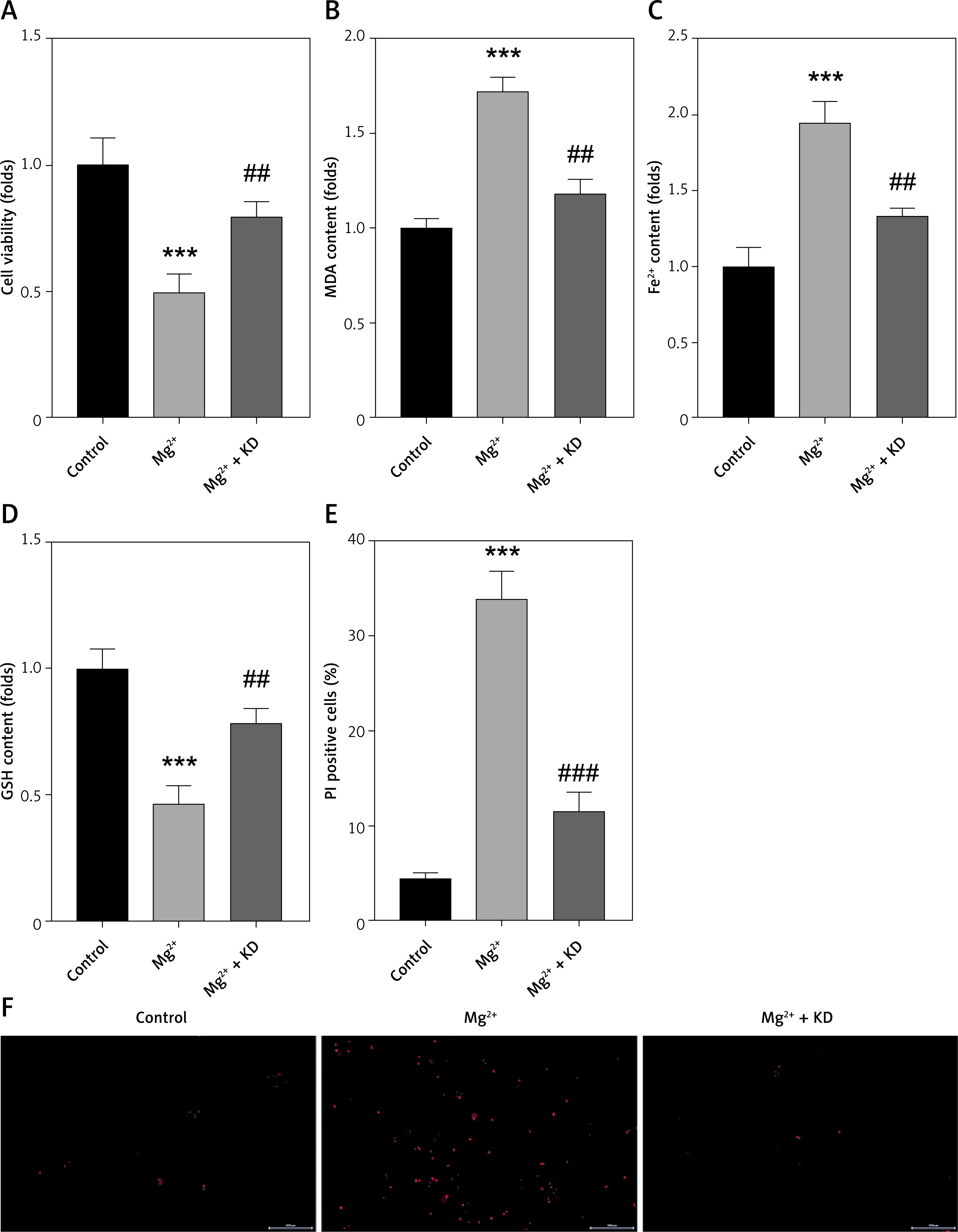
KD suppresses the ferroptosis of neurons induced by epilepsy in vitro
KD is an effective strategy for epilepsy. To conform this, HT22 cells were treated with KD after Mg2+ treatment. We found that the cell viability was significantly decreased by Mg2+ treatment, which was reversed by KD treatment. KD treatment also significantly antagonized the effects of Mg2+ treatment and decreased the release of MDA and ferrous iron (Fe2+), and increased GSH (Figures 2 B–D). KD treatment significantly reduced the percentage of PI-positive cells induced by Mg2+ treatment (Figures 2 E, F). These findings suggested that KD treatment alleviates neuronal ferroptosis in epilepsy.
Figure 2
KD suppresses ferroptosis of neurons induced by epilepsy in vitro. A – Cell viability was detected using CCK-8 assay. B – Release of MDA was determined using MDA assay. C – Release of ferrous iron was detected using ELISA assay. D – Release of GSH was detected using GSH assay. E, F – Death of neurons was determined using PI staining
KD – ketogenic diet, MDA – malondialdehyde (MDA), GSH – glutathione, CCK-8 – Cell Counting Kit-8, PI – propidium iodide. ***P < 0.001, ##p < 0.01, ###p < 0.001.
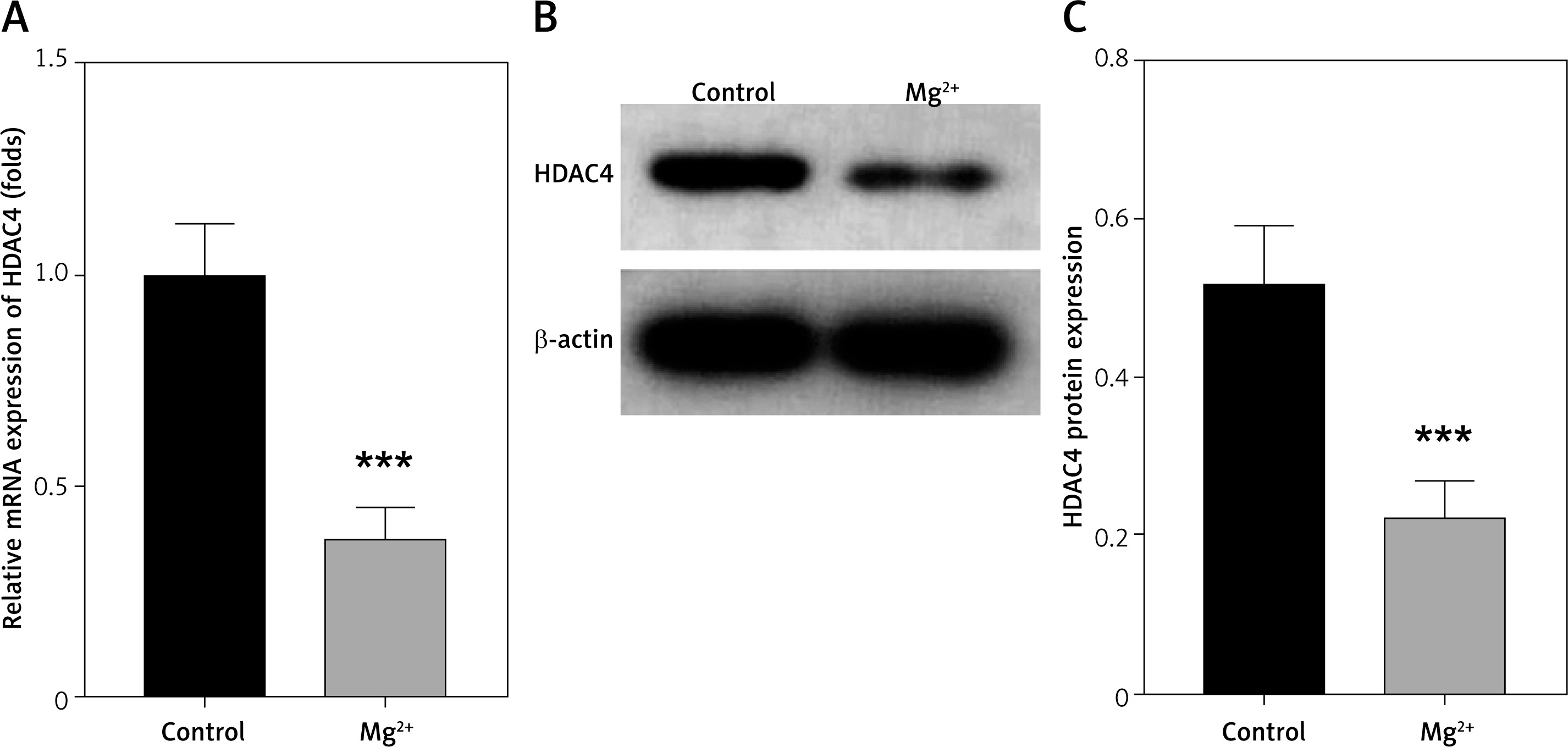
HDAC4 is downregulated in epilepsy
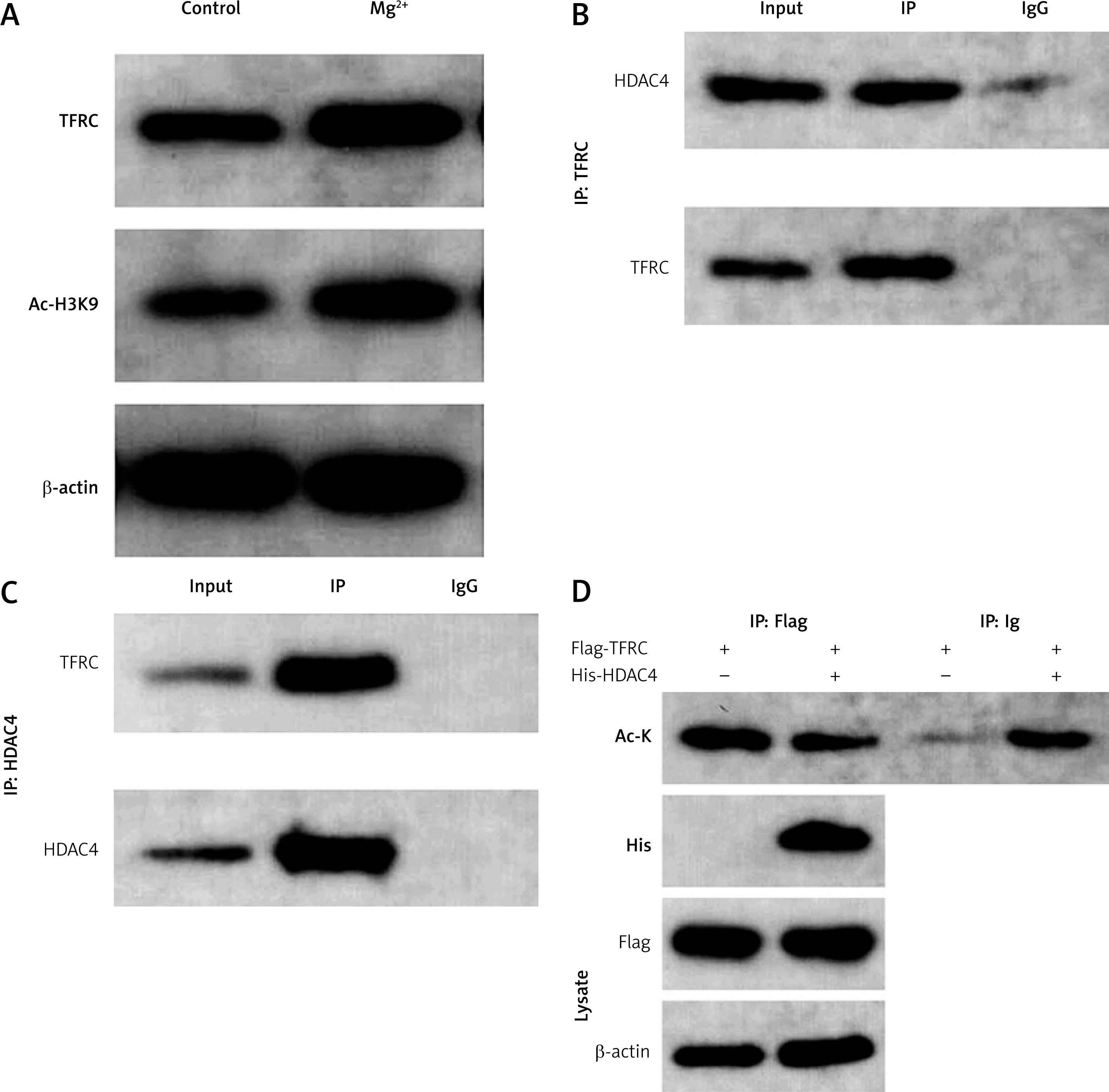
GDS954 was used to analyse genes differentially expressed in epilepsy. We found that HDAC4 was significantly decreased (data not presented here). HDAC4 participates in the progression of epilepsy. Then we determined HDAC4 expression in epilepsy in vitro models. We found that HDAC4 mRNA expression was significantly reduced after Mg2+ treatment (Figure 3 A). This was paralleled by the results from western blot assay. As shown in Figures 3 B, C, Mg2+ treatment significantly suppressed HDAC4 protein expression.
HDAC4 deficiency promotes the ferroptosis of neurons
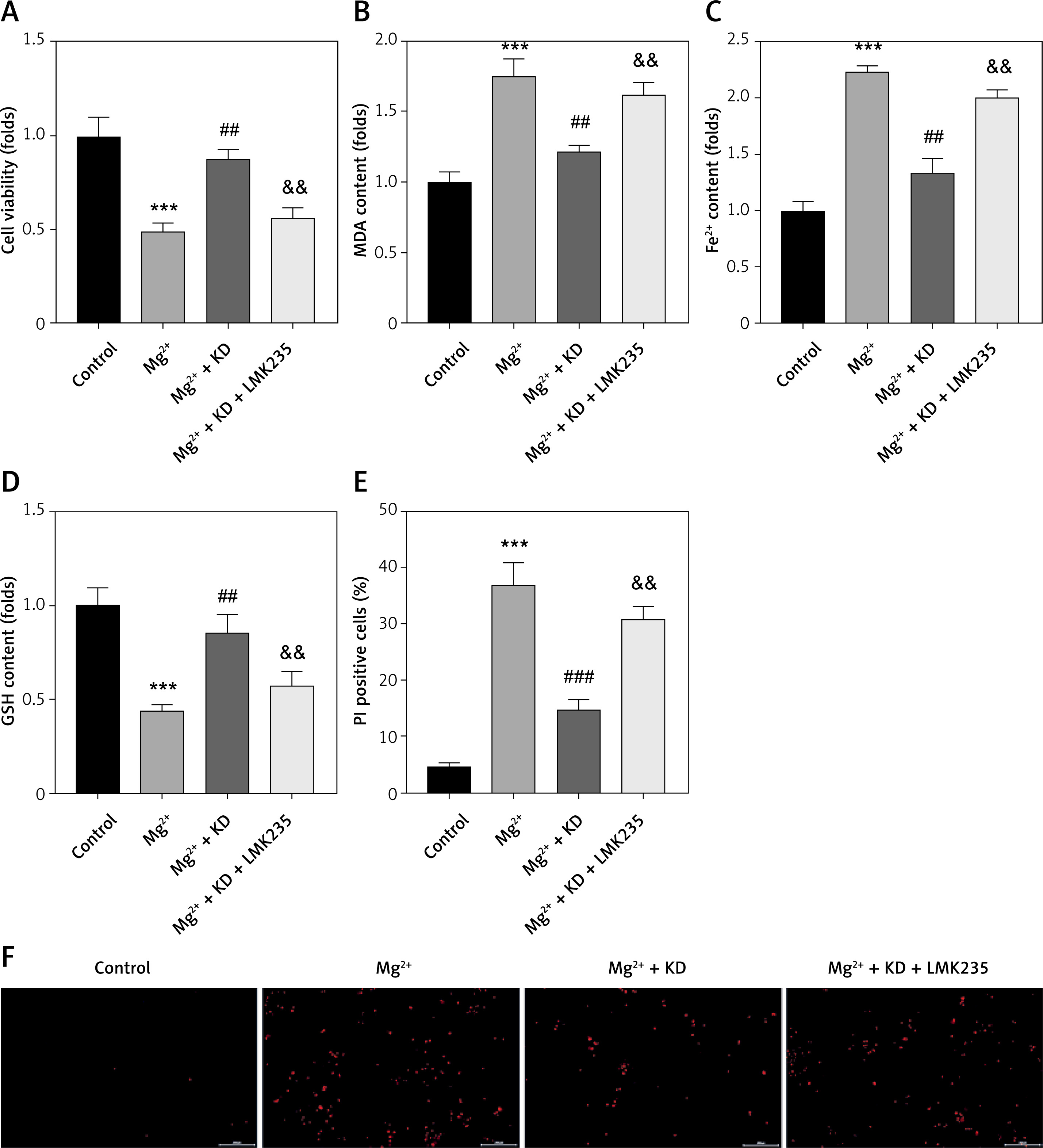
To further confirm the roles of HDAC4 in epilepsy, neurons were treated with the HDAC4 specific inhibitor LMK235. As shown in Figure 4 A, KD treatment-mediated restoration of neuronal viability was significantly alleviated by LMK235. LMK235 also significantly promoted the release of MDA and Fe2+ (Figures 4 B, C), whereas it decreased GSH (Figure 4 D). Moreover, LMK235 treatment significantly antagonized the effects of KD treatment and increased the percentages of PI positive cells (Figures 4 E, F). These findings suggested that KD inhibits the ferroptosis of neurons in epilepsy via upregulating HDAC4.
Figure 4
HDAC4 deficiency promotes ferroptosis of neurons. A – Cell viability was detected using CCK-8 assay. B – Release of MDA was determined using MDA assay. C – Release of ferrous iron was detected using ELISA assay. D – Release of GSH was detected using GSH assay. E, F – Death of neurons was determined using PI staining
KD – ketogenic diet, MDA – malondialdehyde, GSH – glutathione, CCK-8 – cell counting Kit-8, PI – propidium iodide, HDAC4 – histone deacetylase 4. **P < 0.01, ***p < 0.001, ##p < 0.01, ###p < 0.001, &&p < 0.01.
HDAC4 promotes the histone deacetylation of TFRC
Ferroptosis is characterized by iron overload-induced lipid peroxidation [17]. Therefore, we hypothesized that iron metabolism may be involved in the progression of epilepsy. TFRC, as an iron uptaker, promotes the ferroptosis of neurons during the pathogenesis of epilepsy. We found that the protein expression as well as the global acetylation of TFRC was significantly increased after Mg2+ treatment (Figures 5 A, B). HDAC4, as a member of the histone deacetylases, regulates gene expression via exerting histone deacetylase activity. Therefore, we hypothesized that KD may regulate ferroptosis via regulating the HDAC4/TFRC pathway. Endogenous IP assay showed that TFRC can interact with HDAC4 (Figure 5 C). The results of IP experiments showed that overexpression of HDAC4 could significantly reduce the acetylation of TFRC (Figure 5 D).
KD alleviates epilepsy progression in vivo
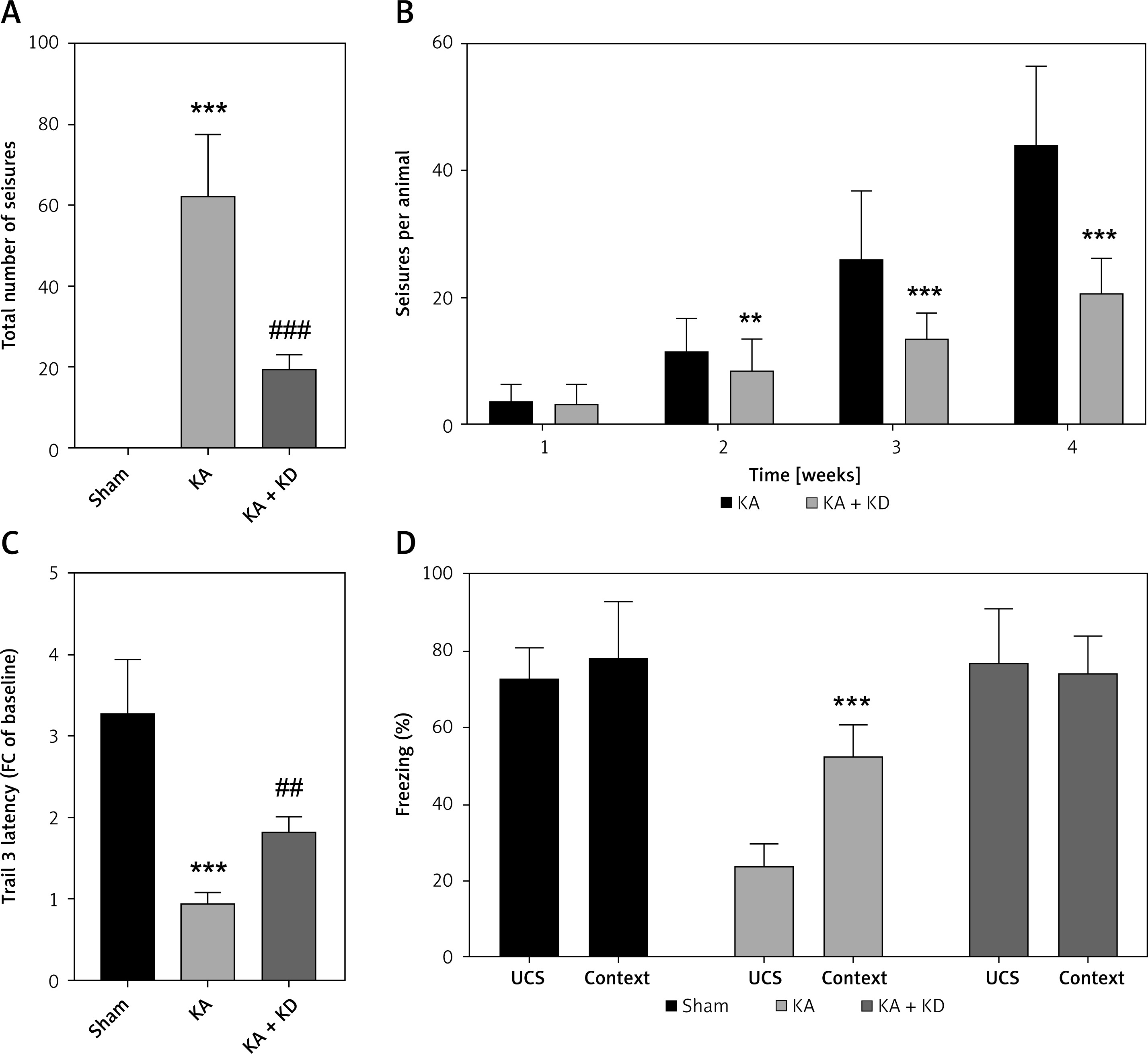
To further confirm the effects of KD on epilepsy, rats were administered KA and/or KD. We found that KD treatment significantly reduced the total number of seizures induced by KA (Figure 6 A). Moreover, KD treatment also significantly decreased the mean number of seizures/week from week 2 (Figure 6 B). We further found that KD treatment significantly improved motor skill learning (Figure 6 C) as well as hippocampal associative memory (Figure 6 D).
Figure 6
Alleviation of epilepsy progression in vivo. A – Total number of seizures after KA and/or KD treatment. B – Mean seizures per week after KA and/or KD treatment. C – Motor learning skill test after KA and/or KD treatment. D – Contextual fear conditioning testing after KA and/or KD treatment
KA – kainite acid, KD – ketogenic diet. **P < 0.01, ***p < 0.001, ##p < 0.01, ###p < 0.001.
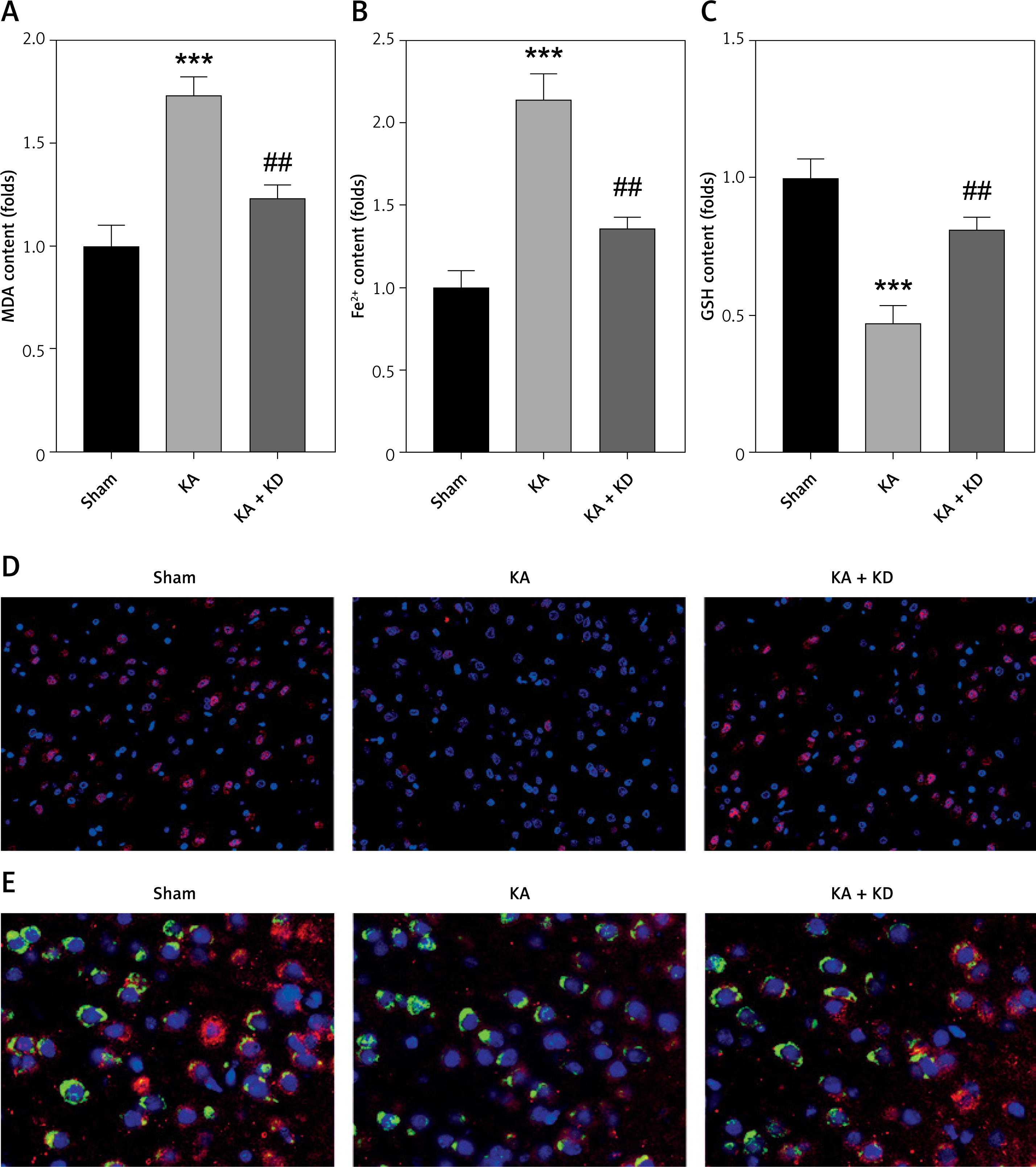
As shown in Figures 7 A–C, KA treatment significantly increased the release of MDA and Fe2+, whereas it decreased GSH; however, this was alleviated by KD administration. KA injection significantly promoted the loss of neurons, which was alleviated by KD administration (Figure 7 D). Moreover, we also found that KD administration reversed the effects of KA treatment and increased the percentages of HDAC4+NeuN+ cells (Figure 7 E). This further confirmed that KD alleviated epilepsy-mediated ferroptosis of neurons via upregulating HDAC4.
Figure 7
KD inhibits ferroptosis in vivo. A – Release of MDA was determined using MDA assay. B – Release of ferrous iron was detected using ELISA assay. C – Release of GSH was detected using GSH assay. D – NeuN expression in brain tissues was determined using immunofluorescence. E – NeuN and HDAC4 expression in brain tissues was determined using immunofluorescence
KA – kainite acid, KD – ketogenic diet, MDA – malondialdehyde (MDA), GSH – glutathione, HDAC4 – histone deacetylase 4. ***P < 0.001, ##p < 0.01.
Discussion
Neuronal loss is a key factor for epilepsy [18, 19]. In this study, KD alleviated epilepsy-induced neuronal ferroptosis. Moreover, KD induced the upregulation of HDAC4. However, HDAC4 deficiency alleviated the effects of KD and promoted neuronal ferroptosis. Additionally, HDAC4 promoted histone deacetylation of TFRC, inducing the downregulation of TFRC. Therefore, KD may protect against epilepsy-mediated ferroptosis of neurons via regulating HDAC4/TFRC signalling.
Accumulating evidence has demonstrated that KD is an effective treatment for epilepsy [7, 20, 21]. KD alleviates drug resistance. Olson et al. [22] reported that KD protects against host metabolism and seizure susceptibility. KD alleviates neurodegeneration and psychiatric disorders [23]. These findings suggest that KD may exert protective effects via modulating metabolism and neuron functions. This study focused on the effects of KD on cellular functions of neurons. We found that KD suppressed iron overload and lipid peroxidation in epilepsy, resulting in restoration of neuron functions as well as inhibition of the ferroptosis-related death of neurons. Epilepsy is accompanied by the loss of neurons. Therefore, KD-mediated inhibition of neuronal ferroptosis can be a promising strategy for epilepsy.
HDAC4 is a key factor underlying brain developmental alterations [24, 25]. For instance, PP1-mediated dephosphorylation and nuclear accumulation of HDAC4 contribute to the neurotoxic effect [26]. Phosphorylation of HDAC4 increases sleep need [27]. Nuclear accumulation of HDAC4 exacerbates cerebral ischemia-reperfusion-induced neuron damage [28]. Interestingly, cytoplasmic HDAC4 recovers synaptic function in Alzheimer’s disease [29]. These findings suggested that the roles of HDAC4 may vary with its location in neurons. In this study, the cytoplasmic expression of HDAC4 was reduced in epilepsy. Moreover, HDAC4 deficiency attenuated the effects of KD and promoted iron overload and lipid peroxidation, contributing to neuronal ferroptosis. Therefore, KD may inhibit neuronal ferroptosis via regulating HDAC4.
HDAC4 participates in regulating biological processes via targeting its downstream effectors. For instance, nuclear HDAC4 interacts with HDAC5 to promote histone deacetylation of Na+/Ca+ exchanger isoform 3 promoter, contributing to neuronal stroke damage [30]. Overexpressed HDAC4 drives histone deacetylation of myocyte enhancer factor 2 and suppresses neural activity in amyotrophic lateral sclerosis [31]. In this study, HDAC4 and TFRC colocalized in the cytoplasm of neurons. Moreover, HDAC4-mediated histone deacetylation of TFRC downregulated the latter expression. TFRC is a key regulator in iron metabolism [32]. However, TFRC is frequently overexpressed in brain disorders, such as stroke, amyotrophic lateral sclerosis, hypoxic-ischemic brain damage, as well as epilepsy [33–36]. Overexpression of TFRC contributes to the continuous accumulation of ferrous iron and autophagic lysosome-mediated degradation of ferritin, and promotes lipid peroxidation and cell collapse, resulting in ferroptosis-related cell death [37, 38]. In this study, TFRC was upregulated in epilepsy. However, KD-mediated overexpression of HDAC4 suppressed TFRC expression as well as neuronal ferroptosis. Although KD may protect against neural loss via inhibiting neuronal apoptosis [23, 39], ferroptosis is differentiated from apoptosis: apoptosis is characterized by the accumulation of apoptosome and executed by caspase cascades, while ferroptosis is characterized by iron overload-induced lipid peroxidation [40, 41]. In this study, increased ferrous iron and lipid peroxidation indicated that the death of neuron in epilepsy was in the form of ferroptosis. Moreover, inhibition of apoptosis, pyroptosis, and necroptosis by its specific inhibitor showed no effects on restoring neuronal function, which confirm the above findings, and further confirmed that epilepsy-mediated neuronal death is in the form of ferroptosis.
Numerous studies have demonstrated the efficacy of the KD in reducing seizure frequency in patients with refractory epilepsy. For instance, 62.5% of children with refractory epilepsy experience a significant reduction in seizures within three days of starting the KD [42]. Noviawaty et al. [43] also observed that KD led to a > 50% reduction in seizure frequency in 45% of adult patients. These findings highlight the potential of the KD as a therapeutic option for epilepsy.
Future clinical trials should aim to further investigate the long-term effects of the KD on seizure control and quality of life in patients with epilepsy. These studies should also explore the potential benefits of the KD in different epilepsy subtypes and patient populations. Mechanistic studies should focus on the detailed pathways involved in KD-induced modulation of neuronal ferroptosis and TFRC signalling. This could involve the use of advanced molecular and cellular techniques to study the effects of the KD on iron metabolism, oxidative stress, and neuronal survival. Research should also explore the potential of combining the KD with other therapeutic interventions, such as antiepileptic drugs or antioxidants, to enhance seizure control and neuroprotection. This could provide a more comprehensive approach to managing epilepsy and improving patient outcomes.
In conclusion, KD protect against epilepsy. KD suppress neuronal ferroptosis in epilepsy via promoting HDAC4-mediated histone deacetylation of TFRC. This may broaden the view of KD in the treatment of epilepsy.