Introduction
Atherosclerosis is a chronic inflammatory disease that usually begins in the first decade of life and is characterised by accumulation of lipids, mononuclear cells, fibrous components and calcium in the arteries [1, 2]. Inflammation can be activated in response to accumulation of cholesterol-rich lipoproteins in the intima layer of the arterial wall, and monocytes and other leukocytes are recruited in the formation of early atherosclerotic lesions [3, 4]. Monocytes mature into tissue macrophages and internalise lipoprotein particles, which over time generate foam cells [4–6].
These and other cells in the vascular wall release multiple inflammatory cytokines into the circulation, such as interleukin-1β (IL-1β), tumour necrosis factor α (TNF-α) and IL-6, which in turn leads to production of C-reactive protein (CRP) in the liver [2, 6]. This elevation of local and systemic levels of inflammatory markers has been associated with increased risk of cardiovascular disease [7]. The most commonly used systemic biomarker of inflammation is high-sensitivity CRP (hsCRP). However, several other biomarkers are also being used and investigated in atherosclerosis, such as high-sensitivity cardiac troponin I, galectin-3, heart-type fatty acid binding protein, and others [8–10].
Despite lifestyle changes and complete adherence to current evidence-based drug therapies, the risk of recurrent cardiovascular adverse events remains during prevention and treatment of cardiovascular diseases [11]. This residual risk is partly due to insufficient reduction of inflammation, and therefore the further development of drugs that target inflammatory pathways represents an important goal for novel therapies in atherosclerosis [11]. The lipid-lowering agents currently in use include statins, ezetimibe, niacin, fibrates and bile acid sequestrants [12]. A further group of novel lipid-modifying agents includes cholesteryl ester transfer protein inhibitors, proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, bempedoic and ethyl eicosapentaenoic acid [13–19], and therapies that target lipoprotein [Lp(a)] [11]. Also, as well as pharmacological approaches, there have been numerous studies that have shown effects of non-pharmacological approaches, such as those involving dietary constituents, on lipid patterns and CRP [20–22]. However, these latter approaches are not the main focus here.
This review is designed to evaluate the effects of different lipid-lowering therapies on inflammatory parameters, with the main focus on CRP. We also discuss the contributions of these therapies to reduced risk of major cardiovascular events.
Effects of lipid-lowering drugs on inflammatory markers
We review here the studies that have evaluated the effects of the different groups of lipid-lowering drugs on high-sensitivity (hs)CRP: statins, ezetimibe, niacin, fibrates, PCSK9 inhibitors and antisense oligonucleotides (ASOs). Table I also provides an overview of the studies discussed here.
Table I
Overview of studies that have evaluated the effects of lipid-lowering drugs on (high-sensitivity) C-reactive protein ([hs]CRP). For full trial names and details, see main text
| Drug | Study | Study population | Treatment | Change in CRP | Primary endpoint | Outcome | Ref. | Level of evidence | |
|---|---|---|---|---|---|---|---|---|---|
| Description | (n) | ||||||||
| Statins | JUPITER | Healthy individuals; LDL < 3.37 mmol/l; hsCRP ≥ 2 mg/l | 17,802 | Rosuvastatin vs. placebo | Rosuvastatin: –47.6% Placebo: –18.6% | Cardiovascular death, myocardial infarction, stroke, hospitalisation for unstable angina or coronary revascularisation | Vascular events: –55% (LDL < 1.8 mmol/l) –62% (hsCRP < 2 mg/l) –65% (LDL < 1.8 mmol/l; hsCRP < 2 mg/l) –79% (LDL < 1.8 mmol/l; hsCRP < 1 mg/l) | [28] | Post-hoc analysis of RT |
| REVERSAL | Coronary stenosis > 20% (angiography); LDL 3.23–5.43 mmol/l | 502 | Atorvastatin vs. pravastatin | Atorvastatin: –3.3% Pravastatin: –35.7% | Change in total atheroma volume | Intensive statin therapy: Greater slowing of rate of progression of atherosclerosis; Greater reduction in LDL and CRP associated with slower rates of progression of atheroma volume | [31] | RT | |
| ATOMIX | Mixed hyperlipidaemia; triglycerides 2.258–5.645 mmol/l; LDL 3.491–6.465 mmol/l | 134 | Atorvastatin vs. bezafibrate | Atorvastatin: –29% to –43% Bezafibrate: –2.3% to –14.6% | Change in CRP (baseline to 1 year) | Atorvastatin: greater reduction in CRP Reductions in CRP directly related to triglyceride changes; inversely related to HDL cholesterol changes | [26] | RT | |
| Ezetimibe | ENHANCE | Familial hypercholesterolemia; LDL > 5.43 mmol/l | 720 | Simvastatin + ezetimibe vs. simvastatin + placebo | Simvastatin + ezetimibe: –49.2% Simvastatin + placebo: –23.5% | Change in carotid-artery intima-media thickness | Combined therapy: greater reductions in LDL and CRP No difference in intima-media thickness changes | [40] | RT |
| IMPROVE-IT | Acute coronary syndrome within preceding 10 days; LDL 1.3–3.2 mmol/l | 18,144 | Simvastatin + ezetimibe vs. simvastatin + placebo | Simvastatin + ezetimibe: –85.0% Simvastatin + placebo: –82.3% | Cardiovascular death, myocardial infarction, stroke, hospitalisation for unstable angina or coronary revascularisation | Combined therapy: greater reductions in LDL and CRP; 2.0% lower incidence of primary end point | [41] | RT | |
| Niacin | Hamoud et al. | Hypercholesterolemia; HDL < 1.03 mmol/l | 14 | Niacin | None | Change in lipid profile, oxidative stress (baseline to 3 months) | Increased HDL and apolipoprotein A1; decreased triglycerides; reduced oxidative stress | [46] | RT |
| Thoenes et al. | Metabolic syndrome | 50 | Niacin vs. placebo | Niacin: –20% Placebo: +1.5% | Change in carotid intima media thickness (52 weeks) | Regression of carotid intima-media thickness; 24% increased HDL; 17% reduced LDL; decreased triglycerides | [47] | RT | |
| Fibrates | FIELD | Type 2 diabetes; cholesterol 2.97–5.43 mmol/l; triglycerides 1.01–4.96 mmol/l or cholesterol/HDL ratio > 4 | 170 | Fenofibrate vs. placebo | Fenofibrate: +38.9% Placebo: –5.9% | Change in carotid intima-media thickness, augmentation index, plasma inflammation markers (baseline to 5 years) | No beneficial changes to carotid intima-media thickness, augmentation index, and plasma levels of inflammation markers | [59] | Post-hoc analysis of RT |
| BIP | Stable coronary heart disease; cholesterol 4.65–6.46 mmol/l; LDL ≤ 4.65 mmol/l; HDL ≤ 1.16 mmol/l; triglycerides ≤ 3.38 mmol/l | 2,979 | Bezafibrate vs. placebo | Bezafibrate: +3.2% Placebo: +3.7% | Change in CRP (baseline to 6.2 years), association with subsequent risk of ischaemic stroke | Each 1.0 natural log unit increase in CRP associated with 37% increased risk of ischaemic stroke | [60] | Post-hoc analysis of RT | |
| DIACOR | Type 2 diabetes, mixed dyslipidaemia | 300 | Simvastatin vs. fenofibrate vs. simvastatin + fenofibrate | Simvastatin: –14.3% Fenofibrate: –27.1% Simvastatin + fenofibrate: –5.4% | Change in hsCRP, Lp-PLA2 (baseline to 3 months) | Combination therapy not more effective Greater reduction in hsCRP and Lp-PLA2 with increased baseline levels | [62] | RT | |
| Agouridis et al. | Mixed dyslipidaemia; LDL > 4.13 mmol/l; triglycerides > 2.26 mmol/l | 90 | Rosuvastatin vs. rosuvastatin + fenofibrate vs. rosuvastatin + omega-3 fatty acids | Rosuvastatin: –53% Rosuvastatin + fenofibrate: –28% Rosuvastatin + omega-3 fatty acids: –23% | Change in non-HDL cholesterol, LDL, Lp-PLA2, hsCRP, plasma isoprostane and paraoxonase (baseline to 3 months) | Reduced non-HDL cholesterol, LDL, hsCRP, Lp-PLA2 for all treatments; greater reductions for rosuvastatin monotherapy | [63] | RT | |
| McKenney et al. (2006) | Mixed dyslipidaemia; LDL 3.36–5.68 mmol/l; triglycerides 2.25–5.64 mmol/l | 576 | Fenofibrate + ezetimibe vs. fenofibrate | Fenofibrate: –21.1% Fenofibrate + ezetimibe: –25.3% | Change in LDL (baseline to week 48) | Similar hsCRP reductions Greater reductions in LDL, HDL, triglycerides, apolipoprotein B for combination therapy | [64] | RT | |
| PCSK9 inhibitors | ODYSSEY COMBO II | High cardiovascular risk; on maximally tolerated statins | 720 | Alirocumab vs. ezetimibe | Alirocumab: –2.1% to –10.9% Ezetimibe: –15.6% to –25.7% | Change in LDL (baseline to week 24) | LDL: Alirocumab: –50.6% Ezetimibe: –20.7% | [69] | RT |
| FOURIER | Cardiovascular disease; LDL ≥ 1.81 mmol/l; on statins | 27,564 | Evolocumab vs. placebo | Evolocumab: –17.6% Placebo: –22.2% | Cardiovascular death, myocardial infarction, stroke, hospitalisation for unstable angina or coronary revascularisation | LDL: –59% Major cardiovascular events: –15% to –20% | [70] | Post-hoc analysis of RT | |
| SPIRE 1, 2 | High cardiovascular risk; stable statins; LDL ≥ 1.81 mmol/l (SPIRE-1); LDL ≥ 2.58 mmol/l (SPIRE-2) | 9,738 | Bococizumab vs. placebo | Bococizumab: –2.1% Placebo: –11.6% | Cardiovascular death, myocardial infarction, stroke, hospitalisation for unstable angina or coronary revascularisation | LDL: –60.5% Major cardiovascular events: –21% (SPIRE-2) | [72] | Post-hoc analysis of RT | |
| ORION-1 | High cardiovascular risk; LDL > 1.8 mmol/l (with history of cardiovascular disease); LDL 2.6 mmol/l (without) | 501 | Inclisiran vs. placebo | Inclisiran: 200 mg single dose: +7.1% 300 mg single dose: –16.2% 500 mg single dose: –19.8% 100 mg two doses: –12.5% 200 mg two doses: –16.3% 300 mg two doses: –16.7%; Placebo single dose: –5.3% Placebo two doses: –20% | Change in LDL (baseline to day 180) | Single dose: LDL: –27.9 to –41.9% PCSK9: –47.9 to –59.3% Two doses: LDL: –35.5 to –52.6% PCSK9: –53.2 to –69.1% | [76] | RT | |
| Bempedoic acid | CLEAR Harmony, CLEAR Wisdom, CLEAR Tranquility, CLEAR Serenity | Hypercholesterolaemia, high cardiovascular risk and stable statins (cohort A); hypercholesterolaemia and statin intolerance (cohort B) | Bempedoic acid vs. placebo | Bempedoic acid: –18.1% (cohort A); –27.4% (cohort B) Placebo: +2.4% to –1.7% | Change in LDL (baseline to week 12) | LDL: –17.8% (cohort A); –24.5% (cohort B) | [78] | Post-hoc analysis of RT | |
| Icosapent ethyl | REDUCE-IT | Triglycerides 1.52–5.63 mmol/l; LDL 1.06–2.59 mmol/l; stable statins; either established cardiovascular disease or diabetes and other risk factors | Icosapent ethyl vs. placebo | Icosapent ethyl: –12.6% Placebo: +29.9% | Cardiovascular death, myocardial infarction, stroke, hospitalisation for unstable angina or coronary revascularisation | LDL: +3.1% Major cardiovascular events: –30% Revascularisation: –34% | [83] | RT | |
| Antisense oligo-nucleotides | Viney et al. (2016) | Lp(a) 125–437 nmol/l (cohort A); Lp(a) ≥ 438 nmol/l (cohort B) | 64 | IONIS-APO(a)Rx vs. placebo | IONIS-APO(a)Rx: –13% to –43% Placebo: –27% | Change in Lp(a) (baseline to day 85, day 99) | Lp(a): –66.8% (cohort A); –71.6% (cohort B) | [84] | RT |
| Tsimikas et al. (2020) | Lp(a) > 150 nmol/l; established cardiovascular disease | 286 | AKCEA-APO(a)-LRx vs. placebo | AKCEA-APO(a)-LRx: 20 mg every 2 weeks: –31% 40 mg every 4 weeks: –30% 20 mg every 2 weeks: –19% 60 mg every 4 weeks: –25% 20 mg every week: –4.5%; Placebo: –33% | Change in Lp(a) (baseline to month 6) | Lp(a): dose-depended reductions | [85] | RT | |
Statins
Statins are competitive inhibitors of 3-hydroxy-3-methyl-glutaryl-coenzyme A (i.e., HMG-CoA) reductase, and they are believed to be the most effective medications for the reduction of plasma cholesterol [23]. The benefits of the use of statins have been shown in large randomised trials, where it has reduced cardiovascular events as both primary and secondary prevention [5, 11, 24, 25]. Although statins have strong hypolipidaemic effects, especially on low-density lipoprotein (LDL) cholesterol, subgroup analyses of large clinical trials (e.g., West of Scotland Coronary Prevention Study (WOSCOPS); Cholesterol and Recurrent Events (CARE); Heart Protection Study (HPS)) have suggested that the beneficial effects of statins appear to extend to mechanisms beyond cholesterol reduction, because of their so-called pleiotropic or cholesterol-independent effects [24, 25]. Many of these effects are mediated through inhibition of isoprenoids, which can improve the function of vascular endothelial cells. This then contributes to the stability of atherosclerotic plaques and reduces oxidative stress and inflammation, thus providing cardioprotective effects [4, 23].
Among the several inflammatory molecules available to monitor inflammation, CRP represents the best-characterised and well-standardised clinical indicator [24]. At least six major prospective studies have supported the hypothesis that elevated CRP levels contribute to increased risk of cardiovascular events: Physicians’ Health Study (PHS); Women’s Health Study (WHS); Atherosclerosis Risk in Communities (ARIC); Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS); Monitoring Trends and Determinants in Cardiovascular Disease (MONICA); and Reykjavik Studies [24, 26]. In particular, Storey et al. studied the relationships between inflammation and cardiovascular disease among patients with chronic kidney disease [27].
The Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER trial) showed that lipid lowering with a statin (e.g., rosuvastatin 20 mg daily) can provide benefits even for individuals with LDL cholesterol levels previously considered acceptable without known atherosclerosis (130 mg/dl; 3.4 mmol/l) but with ongoing subclinical inflammation (CRP > 2 mg/l at baseline) [28]. Here, rosuvastatin reduced median LDL cholesterol by 50% and hsCRP by 37%; indeed, because of the 44% reduction in cumulative incidence of cardiovascular events, the JUPITER trial was stopped after a median follow-up of 1.9 years.
In the Examination of Potential Lipid-Modifying Effects of Rosuvastatin in Combination with Ezetimibe versus Rosuvastatin Alone (EXPLORER) study, 469 patients with hypercholesterolaemia and at high risk of coronary heart disease were treated with rosuvastatin 40 mg alone or in combination with ezetimibe 10 mg, for 6 weeks [29]. The reduction in LDL cholesterol levels was greater with the combination therapy (70% reduction; 189 vs. 57 mg/dl; 4.88 vs. 1.47 mmol/l) compared to monotherapy (57% reduction; 191 vs. 82 mg/dl; 4.93 vs. 2.12 mmol/l) [29]. The combination therapy also resulted in a significantly greater decrease in hsCRP levels (46.4% reduction) versus monotherapy (28.6% reduction) [29]. Also, more patients treated with rosuvastatin plus ezetimibe reached hsCRP levels < 3 mg/l, a level that defines low or moderate risk of future cardiovascular events [29].
The Pravastatin or Atorvastatin Evaluation and Infection Therapy (PROVE-IT) study evaluated intensive (atorvastatin 80 mg daily) versus standard (pravastatin 40 mg daily) treatment strategies with 3,745 patients with acute coronary syndromes, with evaluation of the risk of recurrent myocardial infarction or death from coronary causes [30]. Significantly improved survival was seen for patients who achieved LDL cholesterol < 70 mg/dl (< 1.8 mmol/l) after the statin therapy, and for those with CRP < 2 mg/dl after the statin therapy regardless of their LDL cholesterol levels. Here, the intensive treatment with atorvastatin provided greater lowering of LDL cholesterol and CRP than the standard pravastatin dose. The lowest rate of recurrent events was seen for the patients who achieved concurrent reductions in both LDL cholesterol (< 70 mg/dl; < 1.8 mmol/l) and CRP (< 1 mg/l).
The Reversal of Atherosclerosis with Aggressive Lipid Lowering (REVERSAL) trial investigated the benefits of atorvastatin in terms of reducing atherogenic lipoproteins and CRP levels [31]. The benefits here resembled those of the PROVE-IT study, as the greatest regression of disease was seen for the patients under the more aggressive approach to reduction of LDL cholesterol and CRP.
The Atorvastatin versus Bezafibrate Mixed Hyperlipidaemia Study (ATOMIX) was a 1-year clinical trial in 134 patients with combined hyperlipidaemia [26]. Atorvastatin treatment resulted in a significant 43% decrease in CRP, while patients treated with bezafibrate showed a non-significant reduction. Furthermore, in atorvastatin-treated patients, the reductions in CRP were unrelated to the reductions in LDL cholesterol, although they were directly related to the triglycerides changes and inversely related to the high-density lipoprotein (HDL) cholesterol changes.
The Cholesterol and Recurrent Events (CARE) trial involved 4,159 participants with already established ischaemic heart disease. After 5 years of treatment with pravastatin 40 mg daily, recurrent coronary events were reduced, with a greater effect in patients with higher baseline levels of LDL cholesterol [32]. The changes in CRP levels were measured for 472 randomly selected participants in the CARE trial who remained free of recurrent coronary events during follow-up. Reduced plasma levels of CRP from baseline to 5 years of treatment were observed only in the pravastatin group, and were largely independent of alterations in LDL cholesterol levels [33]. On the other hand, in the placebo group, the CRP levels increased over the 5 years of the trial.
Similarly, in the Pravastatin Inflammation/CRP Evaluation (PRINCE) study, reductions in CRP levels were evaluated [34]. For the 1,702 individuals with no prior history of cardiovascular disease who were enrolled, 837 received placebo; furthermore, together with another 1182 patients with known cardiovascular disease (i.e., secondary prevention), the others under primary prevention received pravastatin 40 mg daily for 24 weeks. In the primary prevention group, baseline CRP was a little lower (0.20 mg/dl) than in the secondary prevention group (0.27 mg/dl). After 24 weeks, reductions in CRP levels were observed in both cohorts treated with pravastatin, as 14.2% for primary prevention and 13.1% for secondary prevention. These reductions in CRP were independent of changes in LDL cholesterol, thus supporting the anti-inflammatory effects of statins [34].
There have been several explanations of the mechanism of the statins for lowering CRP [35]. The first involves mevalonate, which is a precursor of isoprenoid intermediates. Statins inhibit isoprenylation via interference with the phosphorylation of signalling molecules, so the final result is reduced CRP expression in hepatocytes [36]. The second proposed mechanism is suppression of inflammation within the artery, and plaque stabilisation. These can occur via lowering of oxidised LDL, followed by decreased release of inflammatory mediators from atherosclerotic plaques [35]. The results of a very recent in-silico study have also suggested that statins can directly interact with CRP [37]. The final explanation for this lowering of CRP by statins involves genetic factors that are different from the common genetic factors for LDL reduction of cholesterol [38]. Most certainly, different statins lower CRP through different pathways.
Ezetimibe
The lipid-lowering agent ezetimibe selectively inhibits the uptake of cholesterol by the Niemann-Pick C1-like 1 (i.e., NPC1L1) transport protein at the intestinal brush border [11, 25, 39]. This results in reduction of LDL cholesterol, up-regulation of LDL receptors, and increased LDL cholesterol clearance from the plasma [11]. When used in addition to a statin, further reductions in LDL cholesterol, apolipoproteins and triglycerides are seen [11, 39, 40].
The Ezetimibe and Simvastatin in Hypercholesterolaemia Enhances Atherosclerosis Regression (ENHANCE) trial studied the effects of simvastatin and ezetimibe on carotid artery intima-media thickness in patients with familial hypercholesterolaemia [40]. Despite significant reductions in LDL cholesterol (simvastatin, 39.1%; simvastatin plus ezetimibe, 55.6%; p < 0.01) and CRP (23.5% vs. 49.2%, respectively; p < 0.01), the addition of ezetimibe to the highest recommended dose of simvastatin did not provide any further significant differences for the thickness of the carotid artery wall, as compared to simvastatin alone.
On the other hand, in the Improved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT), 18,144 patients who had acute coronary syndrome in the preceding 10 days received simvastatin alone or the combination of simvastatin and ezetimibe [41]. After 7 years of follow-up, the combination of simvastatin and ezetimibe resulted in significantly lower risk of cardiovascular death, major coronary events, or nonfatal stroke than simvastatin monotherapy [41]. Also, significantly more patients treated with the combination achieved LDL cholesterol < 70 mg/dl (< 1.8 mmol/l) and hsCRP < 2 mg at 1 month, in comparison to simvastatin alone. The patients on dual therapy showed improved cardiovascular outcomes [42]. Here, the CRP levels were reduced by 82.3% with the monotherapy, and 85.0% with the combination therapy [41]. Of note, the baseline CRP was > 20 mg/dl, which will be a result of the acute coronary syndrome in the preceding 10 days. Simvastatin alone and in combination with ezetimibe reduced CRP to 3.8 mg/dl and 3.3 mg/dl, respectively [41].
Niacin
Nicotinic acid, or niacin, is a member of the vitamin B family, and it is known to lower total cholesterol, triglycerides and apolipoprotein B-containing lipoproteins (i.e., very-low density lipo protein and LDL), and to increase apolipoprotein A1-containing lipoproteins (i.e., HDL) [11, 43, 44].
As well as its effects on lipid metabolism, niacin is known to be of benefit against cardiovascular risk factors through its inhibition of the generation of reactive oxygen species and of LDL oxidation. This results in inhibition of foam cell formation and monocyte-endothelial cell adhesion and chemotaxis [1, 44]. Lipszyc et al. reported that niacin can reduce IL-1, IL-6 and TNF-α in macrophages, which suggested that it has a role in plaque stabilisation and prevention of progression of atherosclerosis [1].
Interleukin-1, TNF-α, vascular adhesion molecule-1 (VCAM-1) and monocyte chemoattractant protein-1 (MCP-1) are cytokines that are important in the development of atherosclerosis, and they are under the control of nuclear factor-κB (NF-κB) [44, 45]. Through its inhibition of NF-κB activation, niacin inhibits VCAM-1 and MCP-1 expression in endothelial cells induced by TNF-α and IL-1 [44]. This indicates that independent of its effects on lipid metabolism, niacin has vascular anti-inflammatory and potentially anti-atherosclerotic properties [44].
The lipoprotein-independent and potentially anti-atherogenic effects of niacin are mediated by the receptor GPR109A, which is expressed not only in adipocytes but also in monocytes and macrophages [45]. These are key mediators of inflammation, and therefore they represent an important therapeutic target in atherosclerosis [45].
Hamoud et al. studied the effects of niacin in patients with hypercholesterolaemia and low levels of HDL cholesterol [46]. This resulted in significant increases in HDL cholesterol and apolipoprotein A1, and decreased triglycerides (by 23%; p < 0.05) and IL-6 (by 45%; p = 0.044) at 12 weeks [46]. Although serum CRP levels were not affected, there was an inverse linear correlation between serum CRP and HDL cholesterol levels, which illustrated the reduction in oxidative stress of the treated patients [46]. Also, in patients with metabolic syndrome, treatment with extended release niacin 1 g daily for 52 weeks improved endothelial function by 22%, decreased CRP by 20%, increased HDL cholesterol, and decreased LDL cholesterol and triglycerides [47].
In the Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides trial (AIM-HIGH), 3,414 patients with established cardiovascular disease were treated with simvastatin (without or with ezetimibe) to maintain LDL cholesterol at 40–80 mg/dl (1.0–2.0 mmol/l), and then randomly assigned to receive added extended-release niacin or placebo [48]. All eligible patients had low baseline HDL cholesterol (men, < 40 mg/dl (1.0 mmol/l); women, < 50 mg/dl (1.3 mmol/l)), elevated triglycerides (150–400 mg/dl (1.7–4.5 mmol/l)) and LDL cholesterol < 180 mg/dl (4.65 mmol/l) [48]. Despite a significant increase in HDL cholesterol and a decrease in triglycerides, there were no significant differences in cardiovascular events across the two arms, and the trial was stopped after 36 months [48].
The Heart Protection Study 2 – Treatment of HDL to Reduce Incidence of Vascular Events (HPS2-THRIVE) trial included 25,673 patients with vascular disease who received simvastatin (without or with ezetimibe) plus either extended-release niacin plus laropiprant, or placebo [49]. Similarly to AIM-HIGH, there were no significant differences in the incidence of major vascular events across the treatment arms after 3.9 years [49]. Furthermore, the niacin plus laropiprant treatment increased the incidence of diabetes and serious adverse events [49]. Although data regarding the influence of this extended-release niacin on CRP or other inflammatory parameters are not available from AIM-HIGH or HPS2-THRIVE, in patients with stable coronary artery disease, extended-release niacin without laropiprant was shown to significantly decrease CRP levels [50].
Several studies have shown effects of niacin treatment on the levels of the atheroprotective hormone adiponectin [51–53]. The increased plasma adiponectin and the dose-dependent increase in gene expression of adiponectin promoted by niacin treatment indicate another of the pleiotropic roles of niacin, as adipose tissue contributes to systemic and local inflammation associated with atherosclerosis [53].
Fibrates
Fibrates activate peroxisome proliferator-activated receptor (PPAR)-α, and through lowering serum triglycerides and raising HDL cholesterol, they modulate fat metabolism [11, 54]. Along with lipid-modifying properties, fibrates have shown significant reductions in cardiovascular events [54, 55].
Independent of lipid-lowering, fibrates reduce the levels of some inflammatory markers, including CRP, VCAM-1, intercellular adhesion molecule-1 (i.e., ICAM-1) and TNF-α [56, 57]. Through reduction of the nuclear p50–NF-κB–C/EBP-β complex, fibrates down-regulate the expression of CRP in hepatocytes, thus reducing CRP levels [54, 58]. Decreased CRP levels correlate with increased HDL cholesterol, with greater effects seen for patients with dyslipidaemia and higher mean baseline CRP (≥ 3 mg/l) [54, 55].
In contrast, in the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) substudy, after 5 years of treatment with fenofibrate, there were no beneficial changes in inflammation biomarkers in patients with type 2 diabetes [59]. Also, despite reducing recurrent coronary events among patients with high triglycerides or with metabolic syndrome, bezafibrate treatment in the 6-year Bezafibrate Infarction Prevention (BIP) study did not reduce CRP levels [60, 61].
The Diabetes and Combined Lipid Therapy Regimen (DIACOR) trial studied the effects of statin plus fibrate combination therapy on hsCRP in patients with type 2 diabetes, mixed dyslipidaemia, and no history of coronary heart disease [62]. After 12 weeks of daily treatments with simvastatin 20 mg or fenofibrate 160 mg or their combination, hsCRP was significantly reduced from baseline, with the greatest reduction seen for patients with baseline hsCRP > 2.0 mg/l [62]. However, although the statins and fibrates have different mechanisms of action, this combination therapy was not more effective for reduction of hsCRP levels than either simvastatin or fenofibrate alone [62].
Similarly, there were no additional benefits for the combination therapy of rosuvastatin 10 mg and fenofibrate 200 mg versus monotherapy with rosuvastatin 40 mg [63]. Non-HDL cholesterol, LDL cholesterol and hsCRP levels were reduced for both study arms, although the changes were more evident with rosuvastatin monotherapy [63].
On the other hand, fenofibrate 160 mg co-administered with ezetimibe 10 mg for patients with mixed hyperlipidaemia was more efficient for the reduction of LDL cholesterol, triglycerides and non-HDL cholesterol than fenofibrate 160 mg alone [64]. The reductions in hsCRP that were seen for both arms were similar here [64].
PCSK9 inhibitors
The PCSK9 inhibitors represent a new class of lipid-lowering agents. Through their inhibition of PCSK9 degradation of LDL receptors, these human monoclonal antibodies increase LDL receptor activity [11, 55, 65]. When alirocumab, evolocumab and bococizumab were administered subcutaneously as monotherapies or in combination with statins or ezetimibe, they were shown to lower LDL cholesterol levels by 50% to 60% [11, 55, 66, 67]. These PCSK9 inhibitors have also been shown to reduce plasma apolipoprotein B and Lp(a) levels, and therefore have beneficial effects on cardiovascular events [65, 66]. Although inflammation increases PCSK9 liver expression, PCSK9 inhibitors have no significant effects on hsCRP levels, and therefore do not reduce the risk of inflammation [55, 67, 68].
In the 104-week Efficacy and Safety of Alirocumab versus Ezetimibe on Top of Statin in High Cardiovascular Risk Patients with Hypercholesterolaemia (ODYSSEY COMBO II) study, patients with high cardiovascular risk, elevated LDL cholesterol, and on maximum-tolerated statins were treated with subcutaneous alirocumab 75 mg every 2 weeks compared to oral ezetimibe 10 mg daily [69]. After 24 weeks, the reduction in LDL cholesterol was greater with alirocumab (50.6%) than ezetimibe (29.8%) [69]. However, alirocumab reduced hsCRP levels by 10.9% in comparison to 15.6% by ezetimibe, although none of these effects reached significance [69].
The Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Patients with Elevated Risk (FOURIER) trial investigated the effects of evolocumab on prevention of cardiovascular events by baseline hsCRP [70, 71]. Here, 27,564 patients with established atherosclerosis, LDL cholesterol ≥ 70 mg/dl (1.8 mmol/l) and on statin therapy received evolocumab versus placebo for a median of 2.2 years [70, 71]. Compared to placebo, evolocumab reduced LDL cholesterol levels by 59%, with a consequent 20% greater reduction in the risk of major adverse cardiovascular events [70]. Also, evolocumab decreased the inflammatory risk and hsCRP levels (by 17.7%), while the patients with higher baseline hsCRP tended to have greater benefit [70]. In addition, both LDL cholesterol and hsCRP levels were independently associated with adverse cardiovascular events, the rates of which were lowest in the patients with the lowest baseline LDL cholesterol and hsCRP [70].
Similarly, in the Studies of PCSK9 Inhibition and Reduction of Vascular Events trials (SPIRE-1, 2) with bococizumab, there was a connection between elevated hsCRP levels and increased rates of non-fatal myocardial infarction, cardiovascular death and all-cause mortality [72]. Although large reductions in LDL cholesterol were seen for the patients receiving both the statin therapy and bococizumab for 14 weeks, treatment with bococizumab had no effects on hsCRP [72]. Also, this PCSK9 inhibition with bococizumab resulted in the development of high levels of neutralising antidrug antibodies, and therefore these SPIRE trials were closed prematurely [72].
Although these PSCK9 inhibitors had no significant effects on hsCRP levels, these studies show that even with the large reductions in LDL cholesterol seen (i.e., to < 30 mg/dl), CRP remained a risk marker for future cardiovascular events, thus regardless of the LDL cholesterol levels achieved [73]. We can assume that other factors besides LDL cholesterol contributed to the increased inflammation and residual risk. However, in the FOURIER study, the patients with higher hsCRP were seen to benefit most [70].
Inclisiran was the first small interfering (si)RNA to be used for the reduction of proatherogenic lipoprotein cholesterol levels [74]. Although the ultimate aim of all of the drugs from this group is to decrease PCSK9 activity, the mechanism of action of inclisiran is completely different. While evolocumab, alirocumab and bococizumab are antibodies against PCSK9, and thus exert their effects extracellularly, inclisiran inhibits the synthesis of PCSK9 in hepatic tissue. It specifically binds to the PCSK9 messenger RNA (mRNA) in hepatocytes [75], thus targeting both circulating and intracellular PCSK9 in the liver. Whether this difference is clinically important needs to be established in further clinical trials.
In the Trial to Evaluate the Effect of ALN-PCSSC Treatment on Low-Density Lipoprotein Cholesterol (ORION-1) study, the patients were randomly assigned to receive placebo or inclisiran as single doses (inclisiran, 200, 300, 500 mg; day 1) or as two doses (inclisiran, 100, 200, 300 mg; days 1, 90) [76]. Although not reaching significance for the single doses (probably due to the wide distribution), inclisiran 300 mg and 500 mg showed indications of decreased hsCRP (16.2%, 19.8%, respectively). However, patients on the two-dose regime of inclisiran 300 mg showed a significant 16.7% decrease in hsCRP levels.
Taking these trials together, it appears that PCSK9 is not involved in the atherogenic processes with proinflammatory effects.
Bempedoic acid
Bempedoic acid is an oral lipid-lowering prodrug that up-regulates LDL receptor expression and increases LDL cholesterol clearance from the plasma, and it acts through inhibition of adenosine triphosphate-citrate lyase, a key enzyme in the cholesterol synthesis pathway. Activation of bempedoic acid to its active form is achieved by very long-chain acyl-coenzyme A synthetase-1, an enzyme that is expressed in the liver and not in skeletal muscle, but that results in reduced risk of muscle-related adverse effects [77, 78].
Several studies have shown reductions in LDL cholesterol levels by up to 30% with bempedoic acid monotherapy, plus a 24% additional decrease when bempedoic acid was added to stable statin therapy, or a 48% additional decrease in combination with ezetimibe [79–81]. Furthermore, in four phase 3 randomised clinical trials, bempedoic acid provided significant reductions in LDL cholesterol levels: CLEAR Harmony (NCT02666664) [13]; CLEAR Wisdom (NCT029911118) [18]; CLEAR Tranquility (NCT03001076) [82]; and CLEAR Serenity (NCT02988115) [17]. Across all of these studies, a total of 3623 patients received bempedoic acid 180 mg or placebo, once daily for 12 to 52 weeks. After 12 weeks, among the patients with hypercholesterolaemia or high cardiovascular risk, or both, who were on maximally tolerated statin therapy, the mean decrease in LDL cholesterol levels was 17.8%, and for hsCRP levels, 18.1%. A greater treatment effect was seen for the patients with hypercholesterolaemia and history of statin intolerance, with a mean change in LDL cholesterol levels of 24.5% and for hsCRP levels of 27.4% [78].
To determine whether bempedoic acid has cardiovascular protective effects in patients at high vascular risk and with statin intolerance and elevated LDL cholesterol levels, the CLEAR Outcomes study is in progress [15].
Ethyl eicosapentaenoic acid
Ethyl eicosapentaenoic acid (icosapent ethyl) is a highly purified eicosapentaenoic acid ethyl ester that is known to reduce triglyceride levels, and might also have anti-inflammatory, antioxidative, and plaque-stabilising and membrane-stabilising effects [16]. The Reduction of Cardiovascular Events with Icosapent Ethyl-Intervention Trial (REDUCE-IT) involved 8179 patients on stable statin therapy with elevated triglycerides and either established cardiovascular disease or diabetes and other risk factors [83]. After a median of 4.9 years of treatment, icosapent ethyl 4 g daily promoted reduced ischaemic events, less need for revascularisation, and a 12.6% reduction in hsCRP levels [14, 19, 83]. Also, LDL cholesterol levels increased by 3.1% in the icosapent ethyl group compared to 10.2% in the placebo group [83].
Antisense oligonucleotides
Elevated Lp(a) levels represent an independent and highly prevalent risk factor for cardiovascular disease and calcific aortic valve stenosis [84, 85]. ASOs are short synthetic analogues of the natural nucleic acids that bind to target mRNAs via Watson–Crick hybridisation, which results in selective degradation of the mRNA or inhibition of its translation into the protein [86, 87]. Treatment with ASOs that target hepatic apo(a) mRNA have been shown to reduce the plasma levels of Lp(a) [85].
Viney et al. reported on their studies on IONIS-APO(a)Rx and IONIS-APO(a)-LRx. The latter ASO here, IONIS-APO(a)-LRx, is conjugated with a triantennary N-acetyl galactosamine complex and is hepatocyte-directed via an asialoglycoprotein complex [61]. Both of these ASOs have shown not only significant reductions in Lp(a) levels, but also reductions in LDL cholesterol, apoB and oxidised phospholipids [61]. IONIS-APO(a)-LRx is 30 times more potent than its parent ASO, with a mean 92.4% reduction in Lp(a) levels, thus providing lower dosing and enhanced tolerability [84]. Irrespective of coronary artery disease, treatment with IONIS-APO(a)-LRx of patients with Lp(a) levels > 80th percentile and > 99th percentile showed decreased hsCRP of 13% and 43%, respectively. However, these changes were not significant compared to placebo, where hsCRP was decreased by 27%. It needs to be borne in mind here that patients with Lp(a) > 80th percentile had the lowest hsCRP levels, while patients with Lp(a) > 99th percentile had the highest hsCRP levels; hence, the decreases were proportional to the initial hsCRP levels.
Tsimikas et al. studied antisense oligonucleotides further, where they then referred to IONIS-APO(a)-LRx as AKCEA-APO(a)-LRx [85]. In patients with elevated Lp(a) levels and established cardiovascular disease, treatment with AKCEA-APO(a)-LRx resulted in dose-dependent decreases in Lp(a) levels, with the largest reduction of 80% reached at the highest dose of 20 mg weekly [85]. In this study 286 patients with established cardiovascular disease and Lp(a) > 600 mg/l received a saline placebo or AKCEA-APO(a)-LRx as: 20, 40 or 60 mg every 4 weeks; 20 mg every 2 weeks; or 20 mg every week; subcutaneously for 6 to 12 months. The baseline hsCRPs were between 2 mg/l and 3 mg/l across these treatment groups. After 6 months, the absolute mean changes in hsCRP were –0.8 ±5.2 mg/l in the placebo group, and respectively for the 5 active treatments above as –0.9 ±4.2 mg/l, –0.7 ±4.2 mg/l, –0.3 ±2.8 mg/l, –0.5 ±2.2 mg/l and –0.1 ±6.3 mg/l [85]. However, it is not clear whether any of these changes reached statistical significance as hsCRP was measured as a safety outcome, and not as a primary or secondary outcome. Additionally, there were dose-dependent reductions in oxidised phospholipids on apolipoprotein B and apolipoprotein A, which might be important because oxidised phospholipids are responsible for the inflammatory properties of Lp(a). Whether all of these changes are clinically relevant will be answered by the Assessing the Impact of Lipoprotein(a) Lowering with TQJ230 on Major Cardiovascular Events in Patients with Cardiovascular Disease (HORIZON; NCT04023552) trial, which is currently enrolling patients.
Mipomersen is an antisense DNA drug that inhibits apolipoprotein B synthesis, and it has been shown to promote reductions in LDL cholesterol [55, 87]. Although mipomersen improved the overall lipid profile and decreased adverse cardiovascular events, its safety is questionable [88]. Its major side effects included flu-like symptoms, injection site reactions and elevated liver enzymes [55, 88]. Mipomersen therapy has been associated with variable increases in hepatic fat content. The long-term safety of increased hepatic fat content in patients receiving this therapy is uncertain. Mipomersen caused elevated transaminase in some patients, but no cases of severe liver injury have been reported [89]. This might be why data on its effects on inflammatory parameters are scarce at present. In a study by Flaim et al. [90], there was a transient increase in hsCRP shortly after mipomersen subcutaneous injection, but no long-term changes were reported.
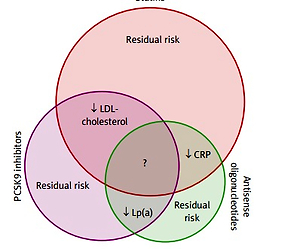
In conclusion, although the rates of morbidity and mortality due to coronary artery disease have been dramatically decreased over the last 30 years, the residual cardiac risk is still substantial (Figure 1). The major, but not only, contributors to reduced morbidity and mortality are the lipid-lowering drugs that generally reduce LDL cholesterol levels. One of the main reasons for the residual risk here is inflammation within the arterial walls, which can be best characterised by the systemic hsCRP levels. Among the lipid-lowering drugs, statins have been shown to decrease cardiovascular morbidity and mortality beyond their LDL cholesterol lowering effect. Other drugs used in secondary prevention of cardiovascular events, such as fibrates and ezetimibe, have less pronounced effects on cardiovascular events. This might be the result of their weaker LDL reduction, as well as their weaker anti-inflammatory effects.
Figure 1
Schematic representation of the known and unknown effects on cardiovascular risk factors of statins, PCSK9 inhibitors and antisense oligonucleotides
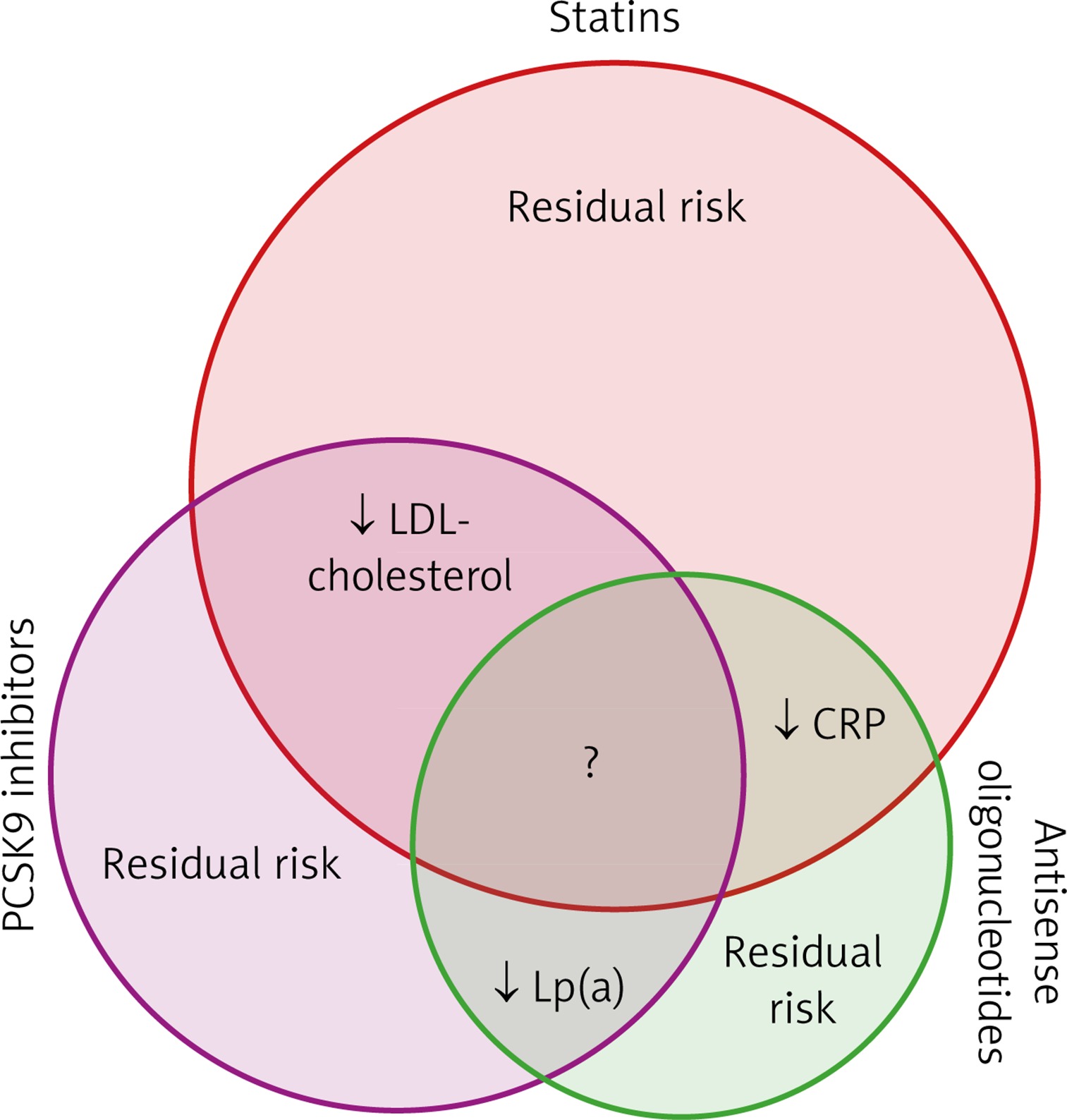
Considering instead the PCSK9 inhibitors, these have little or no effect on inflammatory parameters. However, they can dramatically decrease LDL cholesterol, even in patients treated with the highest doses of statins, and they also further decrease cardiovascular morbidity and mortality. The patients who benefit the most from PCSK9 inhibitors are those with increased baseline hsCRP. Whether these PCSK9 inhibitors have other anti-inflammatory effects than those that reduce CRP levels remains to be determined.
For ASOs, their effects on cardiovascular morbidity and mortality remain largely unknown, although it is known that they have no effects on LDL cholesterol, and that they promote large reductions in Lp(a), and apparently modest reductions in hsCRP. Furthermore, the clinical relevance for some ASOs remains to be determined in terms of their reduction of oxidised phospholipids on apolipoproteins B and A, which are responsible for the inflammatory properties of Lp(a).
It appears that the future direction of lipid-lowering therapies in patients with cardiovascular disease will be for statins without or with ezetimibe to be combined with PCSK9 inhibitors in patients who do not reach their target LDL cholesterol levels. In these patients with elevated Lp(a), the addition of ASOs might also be needed. This last aspect here will become clear with the results of future studies, and in particular with the results of the HORIZON study, which are now fortunately not so far in the future.
From the current perspective of our understanding of the atherosclerotic process, it would be reasonable for future clinical trials that evaluate anti-atherosclerotic drugs to measure their effects on both components, i.e., on lipid markers and on markers of inflammation. Similarly, future strategies in the development of drugs that target atherosclerotic processes should have the dual effects of lowering both lipids and inflammation. However, according to the current knowledge on the mechanisms involved in this lowering of lipids and inflammation, as summarized in the current review, it appears that we are still missing a large piece of the puzzle.