Introduction
Cytokines are molecules with the ability to regulate numerous physiological and pathological processes in living organisms. The number of newly discovered cytokines continues to grow, and the need to systematize them has led to their classification into several basic groups, including interferons, chemokines, hematopoietic cytokines, tumor necrosis factor (TNF) superfamily cytokines, and some growth factors. The basic properties of cytokines are their ability to affect different cell populations in multiple directions (pleiotropism) and redundancy: the ability of different cytokines to exert the same biological effect [1]. The complex pathway of interactions has led to the concept of a “network of cytokines” within which individual elements can act antagonistically, or – by simultaneously affecting effector cells – produce a synergistic effect.
Civilization-related diseases, such as cardiovascular disease and neurodegenerative disorders, present significant therapeutic challenges in the 21st century. Despite numerous clinical observations and experimental studies, our understanding of the pathogenesis of Parkinson’s disease (PD), stroke, and dementia and consequently their treatment, remains inadequate. The inflammatory process, along with pro-inflammatory cytokines, is thought to be a key factor in both the development of vascular lesions and the ensuing neuronal death. Therefore, this study aimed to consolidate existing knowledge about TNF-α in relation to two of the most prevalent civilization diseases: neurodegenerative and cardiovascular conditions. Additionally, this paper highlights the importance of TNF-α in the context of the pathophysiology and treatment of one of the common demyelinating diseases – multiple sclerosis (MS).
The importance of pro-inflammatory cytokines, including TNF-α, in the pathogenesis of many diseases is unquestionable. For instance, TNF-α overexpression has been associated with the clinical figures of various autoimmune and inflammatory disorders, including rheumatoid arthritis, psoriasis, and inflammatory bowel diseases. From a clinical point of view, blocking TNF signaling is currently the most effective therapeutic approach. Therefore, it is difficult to imagine treatment of the diseases listed above without drugs such as infliximab, etanercept, adalimumab, or golimumab [2]. Although inflammation is a recognized residual cardiovascular disease (CVD) risk factor, the knowledge, diagnosis, and management of inflammation-related CVD risk among healthcare professionals remain limited. Nevertheless, they are crucial before targeted therapies become widely available [3].
In accordance with the above, this study aims to systematize knowledge of the synthesis, structure, and molecular regulatory mechanisms mediated by TNF. Given the associated clinical risks, we focus on some of the most common diseases of civilization today: neurodegenerative and cardiovascular diseases. Although the tumor necrosis factor superfamily may not seem to be an obvious choice, it is well established that inflammatory processes underlie the pathogenesis of these conditions.
Molecular basis of TNF
The tumor necrosis factor superfamily (TNFSF) and tumor necrosis factor receptor superfamily (TNFRSF) contain 19 ligands and 29 receptors, respectively. The similar chemical structure of the ligands and the resulting similar affinity with different receptors determine the collective involvement of the proteins in question in the regulation of physiological and pathophysiological processes associated with, among other things, proliferation, activation, and maturation of immune cells as well as apoptosis [4]. One of the most frequently studied representatives of the aforementioned superfamily is TNF. The tumor necrosis factor, which corresponds to the TNF-α form in the majority of published literature, is primarily produced by macrophages and monocytes in response to contact with bacterial cell wall lipopolysaccharides (LPS). The intensity of TNF-α synthesis can be illustrated by the following relationship: during LPS stimulation, the level of mRNA for the TNF gene in macrophages increases 100-fold, while the amount of protein increases up to 10,000 times [5]. For the sake of clarity, it should be noted that tumor necrosis factor β (TNF-β), formerly known as lymphotoxin, is a substance produced by activated lymphocytes. The effects of TNF-β in both physiological and pathological conditions remain poorly understood.
Regulation of TNF gene expression
The TNF gene is rapidly transcribed in a variety of cell types following exposure to a wide range of pathogens and signals of stress or inflammation. Regulation of TNF gene expression at the transcriptional level is cell type and stimulus specific and involves the recruitment of distinct sets of transcription factors to a compact and modular promoter region. The 5′ UTR of the TNF gene contains a proximal promoter region of approximately 200 nucleotides upstream of the mRNA cap site. The TNF gene promoter is sufficient to drive transcription in response to a variety of stimuli, including T cell and B cell activation, calcium ionophore, bacterial LPS, viral infection, Mycobacterium tuberculosis and osmotic stress. TNF mRNA is transcribed within minutes and is independent of de novo protein synthesis. The TNF promoter contains a TATA box and cis-acting elements that are binding motifs for transcription factors: nuclear factor of activated T cells (NFAT), cyclic AMP response element (CRE) and Egr, Ets/Elk, and Sp1 factors. The CRE binding site is a critical regulatory element of the TNF gene in all cell types and under all conditions tested so far [6, 7]. LPS induce TNF expression in macrophages. Several studies have identified signaling pathways that are activated by LPS, including activation of nuclear factor-κB (NF-κB) and activation of mitogen-activated protein kinases (MAPKs), including ERK1 and ERK2, c-Jun N-terminal kinase, and p38. The importance of extracellular signal-regulated kinase1 (ERK1) and ERK2 activation is important for LPS-induced TNF-α production. The ERK signaling pathway is causally involved in the synthesis of TNF-α by human monocytes stimulated with LPS [8–10].
TNF-α is a pleiotropic cytokine. From a chemical perspective, the precursor of the soluble form of TNF is a transmembrane protein with a molecular weight of 26 kDa. In response to a multitude of stimulation signals, this protein is subject to the TNF-α converting enzyme (commonly referred to as TRAY or ADAM 17), which ultimately results in the secretion of the soluble form of TNF-α (sTNF-α) with a molecular weight of 17 kDa [11]. sTNF-α occurs in an active (trimeric) and inactive (monomeric) form.
TNF gene
TNF-α synthesis is modified by genetic factors, especially by variation within the TNF gene. The TNF gene is located on the shorter arm of chromosome 6 at p21.3, in close proximity to the genes of the major histocompatibility complex (MHC) [12]. The TNF gene structure is consistent with that observed in other genes, comprising four exons. The final exon accounts for 80% of the sequence encoding TNF-α. Notably, a portion of the TNF gene sequence is identical to that of other cytokines, including interleukin-1 (IL-1) and interferon-γ (IFN-γ). The promoter region is distinguished by considerable variability. Consequently, the majority of single nucleotide polymorphisms (SNPs) identified in the TNF gene are within its promoter region. Results from experimental model studies and clinical observations suggest that selected SNPs of the TNF gene influence the susceptibility to various human diseases and/or their severity [13–15]. The most studied functional SNPs in the promoter of the TNF gene include -308G >A (rs1800629), -1031T >C (rs799964), -863C >A (rs1800630), and -857C>T (rs1799724) [16].
TNF receptors and their regulation
TNF-α interacts with cells through one of two receptors, currently designated as tumor necrosis factor receptor 1 (TNFR1) or tumor necrosis factor receptor superfamily, member 1a (TNFRSF1A), formerly p55 or p60, and tumor necrosis factor receptor 2 (TNFR2) or tumor necrosis factor receptor superfamily, member 1b, formerly 75 or p80 (TNFRSF1B).
TNFR1, also known as the death receptor, in contrast to TNFR2, which is typical of cells with a nucleus, is expressed on all cell types in the body and has a similar affinity with the membrane and soluble forms of TNF-α. TNFR2, which is mainly found on endothelial cells and immune cells including lymphocytes and macrophages [11], is stimulated by the membrane form of TNF-α. Currently, studies are being carried out to assess the mode of stimulation and functions of each receptor; however, our brief characterization of both receptors suggests that the clinical effects resulting from their stimulation vary. Primarily, TNFR1, in response to the stimulatory signal of TNF-α, promotes cell death and the inflammatory process, whereas binding of TNF to TNFR2 promotes cell survival and tissue regeneration. Interestingly, from a clinical perspective, cell survival is not always beneficial for the patient. Studies have shown that overexpression of TNFR2 in cancer cells leads to tumor growth [17, 18].
TNFR receptor proteins consist of three domains: extracellular, transmembrane, and cytoplasmic. Each of these domains has a different biological role [19]. Both types of receptors, TNFR1 and TNFR2, have cysteine-rich extracellular domains capable of binding a ligand, but their intracellular domains are strikingly different.
The activation of TNFR1 by TNF-α results in the stimulation of four characteristic molecular complexes. The cytoplasmic ‘death domain,’ which is characteristic of TNFR1, recruits the adaptor molecule TNFR1-associated death domain protein (TRADD), which stimulates subsequent signaling proteins such as receptor-interacting serine/threonine-protein kinase 1 (RIPK1), cellular inhibitor of apoptosis protein 1 or 2 (cIAP1/2), TNFR-associated factor 2 or 5 (TRAF2/5), and the linear ubiquitin chain assembly complex (LUBAC). As a result, this signaling pathway stimulates IκB kinase (I kappa B kinase or IKK) activity, which triggers a cascade of processes related to the activation of the NF-κB (nuclear factor kappa B) transcription factor and the MAPK (phosphorylating kinase complex) pathway [2]. This pathway is crucial in stimulating the immune response and inflammation, by affecting, among other things, expression of the genes responsible for synthesizing pro-inflammatory cytokines such as IL-6, IL-1β, and TNF-α itself [20] (Figure 1).
Figure 1
TNF-α signaling pathways. TNF-α binds to the membrane-bound receptors TNFR1 or TNFR2, leading to inflammation, apoptosis, or necroptosis through the formation of signaling complexes I, IIa, IIb, and IIc
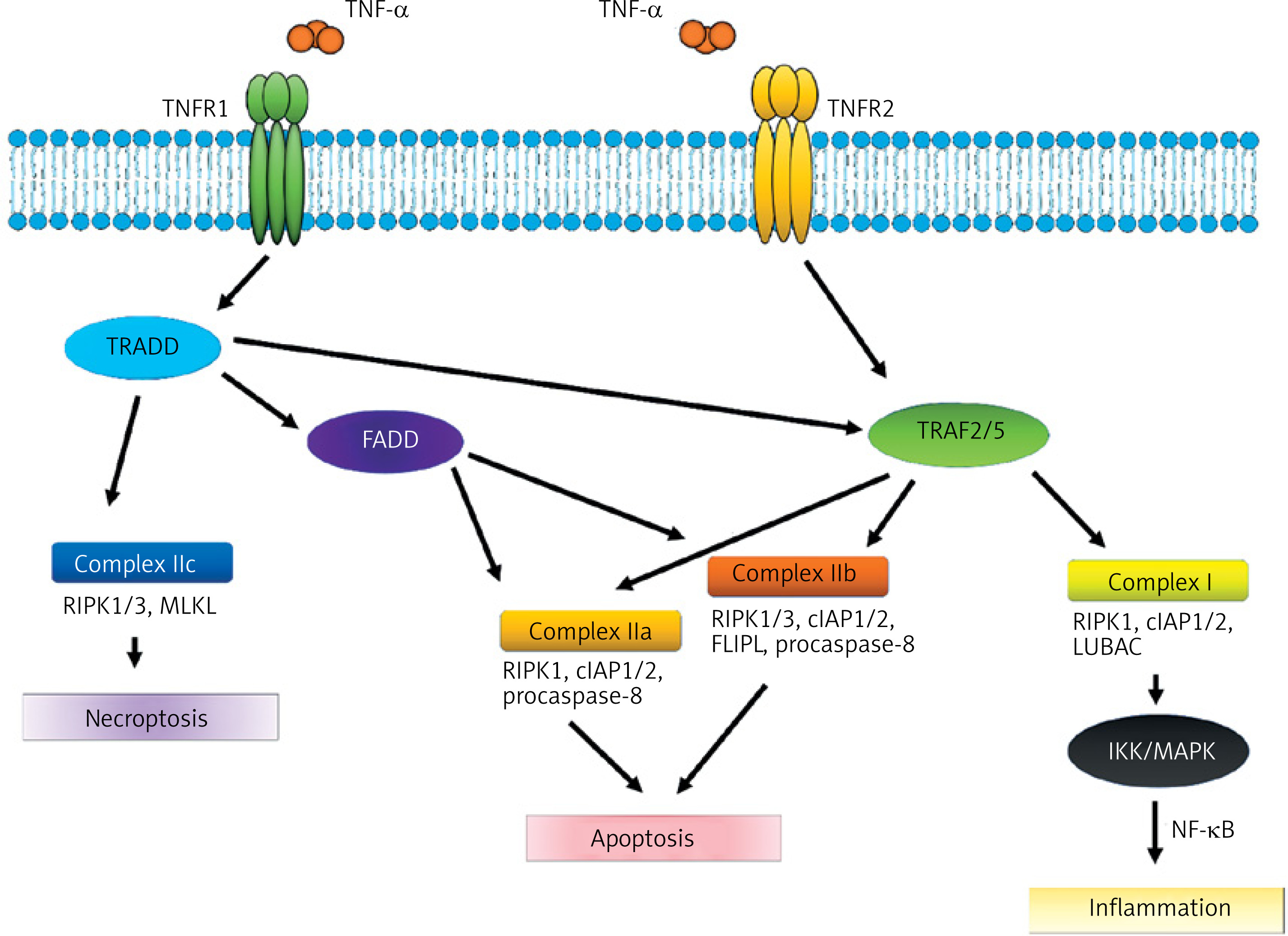
Another complex is divided into two structurally and functionally compatible IIa and IIb systems. The process is analogous and begins with the stimulation of TRADD (TNF-alpha receptor-associated death domain) and Fas-associated protein with death domain (FADD). The remaining components of the aforementioned complex are TNF receptor-associated factor (TRAF2), RIPK1, cIAP1/2 and procaspase-8. Complex IIb is similar to IIa, but additionally contains RIPK3 (receptor-interacting serine/threonine-protein kinase 3). The formation of this complex is regulated independently by the long isoform of FLICE-like inhibitors of protein (FLIPL) that plays a dual role in the activation of caspases and the process of apoptosis. Stimulation of complexes IIa and IIb may also block necrosis. However, the primary effect that they exert appears to be apoptosis (Figure 1).
Another complex, IIc, formed by RIPK1 and RIPK3, stimulates FLIPL-mediated mixed lineage kinase domain-like protein (MLKL), which leads to necroptosis. Apoptosis is different from necroptosis. In the latter, there is a rupture of the plasma membrane, release of intracellular contents into the extracellular space, and, ultimately, a local inflammatory response [21, 22]. TNFR2 lacks a cytoplasmic death domain sequence which is present in FADD. Therefore, the stimulation does not directly result in cell death. As with TNFR1, the impact of combining the transmembrane form of TNF-α with the TNFR2 receptor varies depending on the activated pathways. TNFR2 forms a complex with TNFR, TRAF1, and TRAF2, as well as with cIAP1 and cIAP2. This complex has been observed to interact with the NF-κB inducing kinase (NIK). The function of the κB inhibitor kinase is phosphorylation of IKK. The IKK complex comprises a number of catalytic units that are essential for the correct progression of the phosphorylation process. One potential pathway for NF-κB synthesis involves stimulation of the TNFR2 receptor by TNF-α and NIK activation, which subsequently affects the IKK complex. The IKKβ subunit phosphorylates IκB inhibitors, which results in the activation of NF-κB. This leads to translocation of NF-κB to the cell nucleus, where it binds to specific sequences within the promoter regions of target genes, including A20, cIAP-1, cIAP-2, and Bcl-xL [23, 24]. Furthermore, TNFR2 stimulation may lead to the phosphorylation of endothelial/epithelial protein tyrosine kinase (ETK/BMX), which is associated with vascular endothelial growth factor receptor 2 (VEGFR2). This results in stimulation of the Pl3K/AKT pathway – phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) – which is responsible for regulating cell survival and proliferation.
TNF signaling goal
The outcome of TNFR1-TRADD and TNFR2 signaling is cell survival or death, contingent on the stimulated signaling pathway and cell type. However, the above information suggests that TNFR2 is primarily involved in processes such as proliferation, regeneration, and survival of cells and tissues [25, 26]. Importantly, stimulation of TNFR2 increases the activity of regulatory T lymphocytes and stimulates production of IL-2, which reduces potential autoimmunity and the inflammatory process by inhibiting Th17 cell differentiation. In addition to its effect of stimulating cell survival and proliferation, TNFR2 has also been demonstrated to induce cell apoptosis. This is a consequence of the presence of common elements (e.g. receptors) which are part of two previously described pathways (TRAF and c-IAF) [26, 27]. This fact restricts the potential for using drugs with non-selective affinity for both receptor types.
The role of TNF in the inflammatory process
TNF-α is regarded as a principal cytokine in the acute and chronic phases of the immune and inflammatory responses. As previously stated, the primary stimulus for the synthesis of TNF-α in macrophages is the lipopolysaccharides present in the bacterial cell wall. Furthermore, the factors responsible for the observed increase in TNF-α concentration include INF-γ and IL-1, while IL-4, IL10, PGE2 (prostaglandin E2), and glucocorticoids have been identified as inhibitors of secretion.
The fundamental effect that TNF-α exerts on the immune system is to increase the cytotoxicity of eosinophils, macrophages, and monocytes as well as the phagocytic capacity of neutrophils [2, 28]. Furthermore, the cytokine in question enhances proliferation and differentiation of T lymphocytes, B lymphocytes, and NK cells. Given its integral role within the network of cytokines, TNF-α has the ability to stimulate synthesis of INF-γ, IL-1, and IL-6. Additionally, as a result of autocrine stimulation, TNFα itself undergoes an increase in concentration [29]. Furthermore, TNF-α exerts a direct effect on endothelial cells, leading to the increased expression of adhesion molecules and chemokines that are essential for the accumulation of white blood cells at the site of inflammation. It also induces expression of the molecules in the major histocompatibility complex (MHC I and MHC II).
The onset of acute inflammation gives rise to a cascade of cytokine and chemokine synthesis, attracting immune cells, predominantly neutrophils, to the site of damage. It is now well established that the process of acute inflammation is typically self-limiting, due to the inhibition of pro-inflammatory cytokine synthesis and stimulation of anti-inflammatory protein activity. Inappropriate regulation within the network of cytokines may result in chronicization of the inflammatory process, which may in turn give rise to neurodegenerative processes within the central nervous system (CNS), an elevated risk of carcinogenesis, or cardiovascular complications [16, 30] (Figure 2).
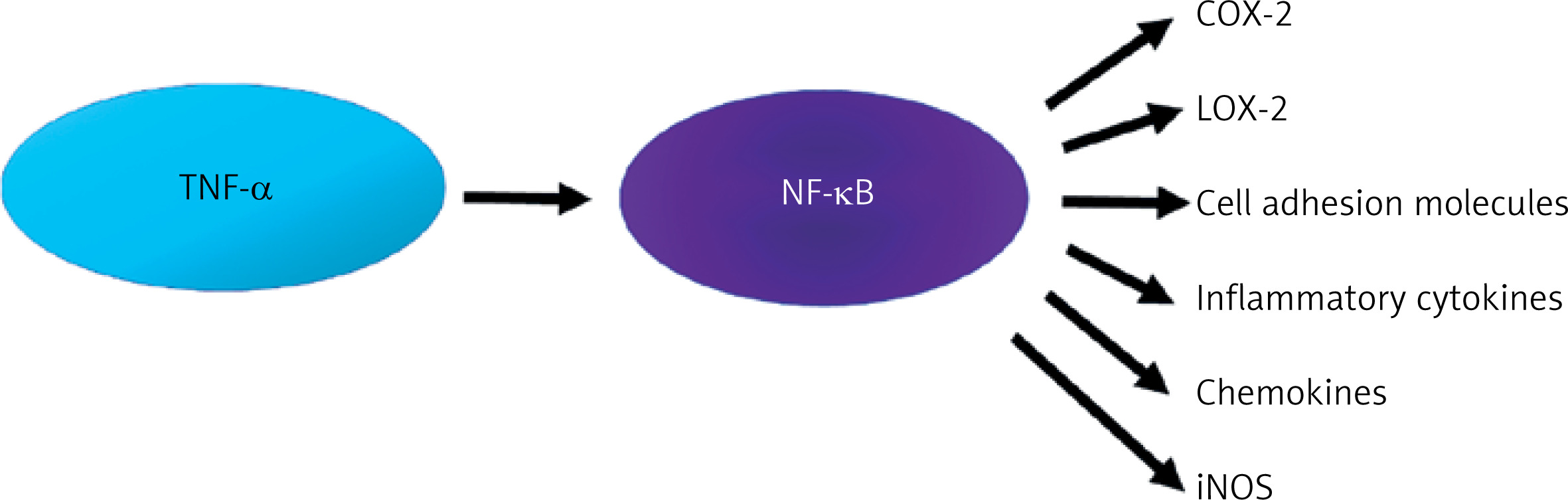
The pro-inflammatory effect of TNF-α is associated with the activation of NF-κB, which is synthesized in almost all cell types. The NF-κB factor is currently the subject of considerable interest due to the possibility of pharmacological modification of its synthesis. For the sake of clarity and coherence, it is necessary to delineate various synthesis pathways, including the canonical and non-canonical NF-κB pathway [31]. It is beyond dispute that NF-κB plays a role in both physiological and pathological processes. Its activity can result in both protection (increased survival, proliferation, synaptic plasticity) and cell damage. Considering mainly the pathophysiological dimension, TNF-α activates NF-κB, which leads to the expression of inflammatory genes responsible for the synthesis of proteins such as cyclooxygenase-2 (COX-2), lipoxygenase-2 (LOX-2), cell adhesion molecules, inflammatory cytokines, chemokines, and inducible nitric oxide synthase (iNOS) [31] (Figure 3).
The association of TNF with diseases of the central nervous system
The central nervous system is the site at which TNF-α both exerts cytoprotective effects and stimulates apoptosis. A significant number of studies currently being conducted using experimental models and clinical observations are primarily focused on the efficacy of drug treatments that aim to reduce the biological effects of TNF-α. TNF-α has been demonstrated to play a role in neuroplasticity and myelination, as well as excitotoxicity, neuroinflammation, and damage to the blood-brain barrier [32–34]. The widespread use of drugs that block the synthesis of TNF-α limits the pleiotropic effects of this cytokine. Although anti-TNF-α therapy has been successfully introduced in the treatment of neurosarcoidosis, it has been observed that blocking its synthesis in patients with MS may, paradoxically, result in exacerbation of the disease. There is evidence that that intravenous treatment of MS patients with the anti-TNF antibody may lead to intrathecal immune activation and may therefore be harmful for MS patients [35, 36]. Similarly, administration of TNF-α in experimental autoimmune encephalomyelitis (EAE) mice has been shown to exacerbate the course of the disease [20].
TNF-α should be treated as a cause of neurological diseases. The results indicate that TNF-α induces neurological dysfunction. TNF-α-induced brain dysfunction and disease are caused by necroptosis and microglial activation in hippocampal neurons. TNF-α is an important cytokine involved in various homeostatic and pathogenic bioactivities [37]. The dualistic nature of TNF-α is particularly evident in the central nervous system. As detailed in Table I, this cytokine is involved in both inflammatory processes and cell death. It also plays a key role in maintaining homeostasis of the central nervous system by affecting glial cell proliferation and neuronal function. TNF-α plays a special role in regulating survival of oligodendrocyte cells as well as formation and repair of myelin. This explains the limited use of drugs that reduce synthesis or the biological activity of TNF-α in patients with multiple sclerosis.
Table I
Biological effects of TNF-α on the central nervous system
Parkinson’s disease
Parkinson’s disease (PD) is the second most common neurodegenerative disease after Alzheimer’s disease (AD). The estimated prevalence of PD is 0.3% in the general population and 1.0% and 3.0%, respectively, in people older than 60 and 80. A special group of patients is those with early-onset Parkinson’s disease (EOPD), defined as PD with age at onset (AAO) after age 21 years but before the typical AAO for PD. Consensus is lacking, and the reported maximal age for EOPD has varied from 40 to 60 years [38]. The etiology of PD is not well established. For several decades, environmental factors were considered primarily responsible for disease development. However, recent studies have demonstrated that PD has a substantial genetic component, probably causing or influencing at least 10% to 25% of cases, more commonly in younger patients. Therefore, the new concept of PD as an “eco-genetic” disease assumes an influence of environmental and genetic factors coinciding with the aging process [39]. Indeed, a family history of PD is reported in 20% of patients with EOPD versus 6.9% of patients with later onset PD, and the age-specific risk of PD is 7.8-fold higher in the relatives of patients with EOPD compared with 2.9-fold among the relatives of patients with later onset PD [38]. Recent findings have identified inflammation as a potential factor contributing to PD development. It was hypothesized that genetically determined differences in immune system function, especially anti- and pro-inflammatory cytokine production, might influence the risk of sporadic PD development and/or onset [40, 41]. Of particular interest within the group of pro-inflammatory cytokines is TNF-α. The involvement of TNF-α in the etiology of PD, both in its early and late stages, has been substantiated using both experimental models and clinical studies [42]. TNF-α and TNFR receptors are responsible for the activation of two independently running pathways. As previously stated, each can result in apoptosis or an increase in the expression of numerous anti-apoptotic genes [43]. In PD, the negative effect of the cytokine on the survival and function of neurons appears to be predominant. The results of studies using experimental models showed a significant increase in the concentration of mRNA for TNF-α in the substantia nigra (SN) of rats treated in vivo with the most commonly used experimental neurotoxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) [44]. TNF-α not only induced inflammatory responses but was also involved in the maintenance of the inflammatory process that had already been initiated. This is demonstrated by the sustained elevation of the cytokine in question one year following the administration of the neurotoxin. One of the mechanisms proposed as a potential cause of the neurodegeneration characteristic of parkinsonism is the impact of TNF-α on the functions of microglia. TNF-α activates microglia, which leads to a progressive loss of dopaminergic neurons in the SN [45]. As shown by clinical studies, low concentrations of TNF-α in the brains of healthy adults are observed in contrast to high concentrations of TNF-α and the soluble form of the TNFR1 receptor in the cerebrospinal fluid as well as the SN obtained from the brains of patients diagnosed with PD [44–46]. Dopamine neurons in the substantia nigra of the brain exhibit a high degree of sensitivity to the effects of TNF-α. TNF-α can activate microglia in the interbrain, stimulate the inflammatory response, and exacerbate oxidative stress through ROS- and NO-dependent mechanisms.
Elevated levels of TNF-α have been detected in the substantia nigra of patients with PD and atypical parkinsonism. Elevated levels of TNF-α have also been found in the cerebrospinal fluid of people with REM sleep behavior disorder [45, 47, 48]. This is a particularly interesting observation given that rapid eye movement sleep behavior disorder (RBD) is considered a non-motor manifestation of prodromal synucleinopathy [49]. TNF-α levels may also influence the progression of the disease. Menza et al. reported that TNF-α levels were positively correlated with cognitive impairment, depression, and disability in patients with PD [50]. Furthermore, Wang et al. investigated the correlation between plasma levels of inflammatory cytokines as well as alpha-synuclein (α-syn) and fatigue in patients with PD. The study included 63 patients with PD, 35 patients with symptoms of fatigue and 28 patients without clinical symptoms of fatigue. In addition to factors related to disease progression, stage, and older age, the authors demonstrated that plasma levels of the cytokines IL-1β, IL-18, TNF-α, and phosphorylated α-syn (p-α-syn) were positively correlated with the results on the Fatigue Severity Scale 9 (FSS-9). Furthermore, a higher score on the Parkinson’s Fatigue Scale 16 (PFS-16) was associated with elevated levels of TNF-α and p-α-syn in plasma. The findings suggest a potential correlation between elevated TNF-α and p-α-syn levels in plasma and the onset of fatigue in individuals with PD [51].
The confirmation of the relationship between TNF-α and the risk of PD, as well as the progression of the disease, presents an opportunity for pharmacotherapy with drugs that block the activity of the cytokine in question. The findings of the research studies that have been published to date are encouraging. For example, Peter et al. found that the use of TNF inhibitors was associated with a 78% reduction in the likelihood of PD (incidence rate 0.22; 95% CI: 0.05, 0.88) [52]. The study was conducted on a group of 144,018 individuals diagnosed with inflammatory bowel disease (IBD), matched for age and sex with a control group of individuals unaffected by IBD, who were treated with adalimumab, certolizumab, golimumab, and infliximab.
The pathogenesis of diseases of the central nervous system, including PD, is a complex process. In addition to environmental factors, there is evidence to suggest that genetic predisposition also affects the risk and progression of PD. Results from genetic studies investigating the variability of genes encoding TNF-α and TNFR activity remain inconclusive [53, 54]. Although there are isolated reports of neuroprotective effects associated with high concentrations of TNF-α, the cytokine generally exerts an opposing effect to that of IL-10 in the majority of processes occurring in the human body. Genetically determined inter-individual differences in cytokine production may be at the root of differences in the progression of the inflammatory process [55, 56]. TNF gene polymorphism, specifically the -308G >A variant, which has been most extensively studied in the Caucasian population, is associated with differences in the expression of the encoded protein. The -308A variant allele increases the transcriptional activity of the gene compared to the more common G allele, which may clinically correspond to the severity or dysregulation of inflammatory responses. The study by Nishimura et al. on a group of patients with sporadic PD showed that SNP -1031T>C, -863C >A and -857C>T may influence the risk and age of developing PD in oriental populations [56]. In patients with EOPD, the frequency of the -1031C allele, which is associated with increased transcriptional activity and TNF-α concentration, was significantly higher compared to patients with late-onset Parkinson’s disease (LOPD) and healthy subjects. Furthermore, the -1031C allele and the -1031CC genotype were identified as risk factors for PD [5]. It is noteworthy that the -1031C allele, which is associated with increased TNF-α synthesis in oriental population patients, corresponds phenotypically to the -308A variant of the -308G >A polymorphism, which is prevalent in the Caucasian population.
Subsequent research by Kang et al. investigated similar issues, focusing on the genetic variability of TNFRSF1A, which is responsible for the expression of the TNFR1 receptor [57]. The aim of the study was to assess the association between selected genetic variants of the gene encoding the TNFR1 receptor and the development of PD. Previous research had linked the effect of pharmacological blockade of TNFR1 signaling during anti-TNF therapy to genetically determined TNFRSF1A variability. Using genome-wide association study (GWAS) data from the International Parkinson’s Disease Genomics Consortium and 23andMe, Inc, the effects of TNF-TNFR1 inhibition on PD risk and age at onset of PD were investigated in relation to genetic variants associated with lower levels of circulating C-reactive protein. Despite the large number of controls analyzed, the importance of inhibiting TNF-TNFR1 signaling in preventing or delaying the onset of PD symptoms was not confirmed.
Alzheimer’s dementia
Alzheimer’s disease (AD) is one of the most common forms of dementia. A typical pathological feature of AD is the accumulation of β-amyloid (Aβ) plaques and neurofibrillary tangles. The pathogenetic model under consideration fails to account for a substantial proportion of the clinical and pathological variability observed in the disease. Consequently, further hypotheses have been formulated, of which one suggests that chronic inflammation within the CNS may be a significant contributing factor to the development and progression of the disease [58]. Hypotheses of AD pathophysiology also include amyloid cascade, inflammation, vascular, and infection factors [59, 60].
In the CNS, TNF-α is secreted by various cell types, mainly microglia. Due to the complex interactions of TNF-α with other cytokines, this factor plays a key role in the regulation of numerous physiological and pathophysiological changes in the brain. Primarily, TNF-α exerts an effect of increasing synaptic transmission by both direct and indirect means, including the additional release of glutamate from astrocytes. By activating pathways associated with NF-κB, it is involved in regulating neuronal function in the key areas of the brain that regulate sleep [61]. TNF-α is a major pro-inflammatory cytokine that stimulates production of IL-1, IL-6, and IL-8, promoting chronic inflammation, especially when the production of pro-inflammatory cytokines is not balanced by anti-inflammatory cytokines such as IL10 [62].
The importance of TNF-α in the pathogenesis of AD has been confirmed in clinical studies and experimental models, in the context of both beta-amyloid and tau protein [63]. Experimental studies have shown that TNF-α increases the expression of myeloid precursor protein (APP) and β-site amyloid precursor protein cleaving enzyme-1 (BACE1) in mouse astrocytes. Furthermore, it has been demonstrated that it stimulates the activity of γ-secretase in HEK cells, thus releasing Aβ peptides [64]. The splitting of microglia, a phenomenon observed in dementia, has been shown to facilitate production of TNF-α. This, in turn, sustains the persistence of chronic inflammation, thereby stimulating the synthesis of Aβ and subsequently contributing to the loss of neurons. Recently, a new concept has emerged linking the triggering receptor expressed on myeloid cells 2 (TREM2) to the role of TNF-α in the pathology of AD. TREM2 is found in various tissue macrophages, such as CNS microglia, bone osteoclasts, alveolar, peritoneal, and intestinal macrophages. TREM2 is also present on cultured bone marrow-derived macrophages and monocyte-derived dendritic cells. Some researchers have identified a rare variant of TREM2 that is a risk factor for AD [65]. TREM2 has been reported to bind to proteins with a strong tendency to aggregate and accumulate in neurodegenerative diseases, such as β-amyloid and supports the microglial response to Aβ plaques [66]. Guerreiro et al. and Jonsson et al. demonstrated the association between variability in the gene encoding TREM2 expression and the risk of late-onset sporadic AD. The authors indicated that heterozygous hypomorphic variants of TREM2, mainly TREM2R47H, increase the risk of AD [67, 68]. Impaired receptor function is responsible for the limited ability of microglia to encase Aβ plaques, leading to the spread of β-amyloid accumulation, severity of neurotoxicity, and the progression of the disease. At present, studies are being carried out to assess the efficacy of TREM2 agonists in the treatment of AD [69].
Clinical studies of patients with mild cognitive impairment (MCI) showed higher levels of TNF-α and tau protein and lower levels of Aβ42 and the anti-inflammatory cytokine transforming growth factor β (TGF-β) in patients with MCI compared to the control group [70]. In addition, the authors observed an association between elevated levels of TNF-α in the cerebrospinal fluid and the conversion of MCI to AD. However, this observation was not confirmed in recent research by Fu et al. [71].
Clinical evidence for the importance of TNF-α in the pathogenesis of CNS diseases, including AD, is provided by observational studies of patients with systemic inflammatory diseases treated with TNF-α inhibitors [72, 73]. Zhou et al. published the results of a retrospective clinical study based on the medical records of 56 million adult patients [72]. The aims of the study were to investigate the association between inflammatory diseases and AD dementia and to determine whether the drugs that block tumor necrosis factor functions, used in patients with rheumatoid arthritis (RA), psoriasis and other inflammatory diseases, affect the risk of AD. The study demonstrated an elevated susceptibility to AD development among the study group of patients, with those diagnosed with RA, inflammatory bowel disease, or Crohn’s disease exhibiting the highest risk (p < 0.0001). Importantly, the use of drugs that target TNF-α may influence the development of dementia. Etanercept, adalimumab, and infliximab were associated with a significantly lower risk of AD in patients with RA. In patients with psoriasis, this association was only confirmed for etanercept and adalimumab. This study may provide a basis for further clinical trials of drugs in this class, with a potentially protective mechanism of action, particularly in patients with inflammatory diseases who are predisposed to developing dementia.
The current evidence suggests that the clinical efficacy of drugs in this class is more significant in patients with inflammatory conditions. For example, Chen et al. found cognitive improvement in elderly patients with RA following the administration of etanercept and adalimumab. Conversely, etanercept exerted a minimal impact on the core symptoms in patients with mild and moderate AD [74, 75]. There is little interest in the use of this group of drugs, and currently no clinical trials are being conducted [https://clinicaltrials.gov/]. This may be related to the pharmacokinetic properties of this class of drugs. The limited efficacy of subcutaneous anti-TNF-α therapies for AD may be attributed to the high molecular weight of anti-TNF-α monoclonal antibodies, which presents a challenge in their crossing the blood-brain barrier under physiological conditions.
Neuroinflammatory diseases
Neuroinflammation is a common pathological characteristic of several CNS disorders. It is characterized by the activation of glial cells, demyelination, and neurodegeneration. In autoimmune diseases of the CNS, TNF-α is a key inflammatory cytokine that regulates peripheral and local immune responses. Its involvement in excitotoxicity and neuroinflammation highlights its impact on CNS pathophysiology and emphasizes its multifaceted roles in regulating cellular responses and inflammatory processes within the CNS. Neuroinflammatory diseases encompass a range of conditions, including neurosarcoidosis, neuro-Behçet’s disease – a systemic inflammatory vasculitis of unknown origin – and MS. However, given the breadth of this subject, this article will focus solely on multiple sclerosis.
Multiple sclerosis
MS is an autoimmune demyelinating and neurodegenerative disease of the CNS. The most well-known concept of MS is based on the co-occurrence of genetic susceptibility and a specific immune system response against its own antigens, which occurs in response to environmental factors. The signal that initiates demyelination is the activation of CD4+ T lymphocytes in response to myelin antigens. Myelin proteins are presented by antigen-presenting cells (APC) in combination with the MHC class II antigens. Subsequent steps involve the passage of autoreactive T lymphocytes to the CNS and differentiation into subclasses showing pro-inflammatory (Th1 and Th17) or anti-inflammatory (Th2) activity [76, 77]. Proinflammatory cytokines, including TNF-α, IFN-γ, IL-2, IL-17, are involved in the activation of microglia and B lymphocytes, as well as the influx and stimulation of peripheral monocytes and macrophages. This briefly outlined pathway of pathophysiological changes indicates the importance of cytokines, including TNF-α, in the initiation and propagation of the autoimmune process that characterizes demyelination in the progression of MS.
Clinical studies of patients with multiple sclerosis have shown a correlation between both the severity of symptoms and the progression of the disease and elevated levels of TNF-α in the cerebrospinal fluid [78]. However, a double-blind, placebo-controlled phase 2 study of 168 patients with relapsing-remitting MS treated with lenercept showed exacerbation of the disease. The study revealed that patients treated with lenercept (p55 tumor necrosis factor receptor fusion protein) exhibited a significantly higher incidence of disease exacerbation at the 48-week mark compared to those receiving placebo (p = 0.007). However, treatment had no influence on the Expanded Disability Status Scale (EDSS) score in either group after 24 weeks of treatment and at the final clinical assessment [79]. The explanation for the above observation should be sought in the results of studies conducted using animal models. It has been suggested that chronic inhibition of TNF-α may enhance the T lymphocyte response to a specific antigen. Paradoxically, drugs that inhibit TNF-α activity may enhance the pro-inflammatory activity in the CNS as well as increasing survival of peripheral myelin-autoreactive T lymphocytes. Furthermore, drugs based on this mechanism of action may result in a reduction of IL-10 levels and an increase in IL12 and IFN-c, thereby creating a profile comparable to that observed in patients with multiple sclerosis [80, 81]. Another theory is that the distribution of TNF-α levels between the CNS and peripheral tissues is dysregulated (higher levels in the CNS due to the impermeability of the blood-brain barrier). The non-selective inhibition of TNF-α activity by affecting two receptors (TNFR1 and 2) may be considered potentially harmful for patients with multiple sclerosis. This is also confirmed by a study conducted on a group of patients with rheumatic diseases in whom anti-TNF-α therapy resulted in the occurrence of demyelinating lesions [82].
Drugs that non-selectively inhibit TNF-α binding to the receptor and inactivate its effect limit transmembrane interaction with both TNFR1 and TNFR2. As mentioned above, TNFR2 plays an important role in the proliferation of immature oligodendrocytes, remyelination, and inhibition of autoreactive CD4+ T lymphocytes. Although some studies have questioned the role of anti-TNF-α drugs in the demyelinating process [82, 83], until drugs that selectively block TNFR1 activity are developed, drugs in this group should be used with great caution, especially in the case of MS comorbidity with rheumatological diseases. Another issue is the risk of inflammatory central nervous system diseases in patients receiving anti-TNF therapies. The results of a systematic review and meta-analysis of 18 studies, including more than 1 million patients with autoimmune diseases, were published by Xie et al. recently [84]. The incidence rates of new-onset inflammatory CNS events after initiating anti-TNF therapies ranged from 2.0 to 13.4 per 10,000 person-years, and overall, exposure to this treatment was associated with a 36% increased risk of any inflammatory CNS disease compared to conventional therapies (RR = 1.36; 95% CI: 1.01–1.84; I2 = 49%) with similar risk across different types of autoimmune diseases and TNF inhibitors. The role of TNF-α in neurology diseases is summarised in Table II.
Table II
Summary of selected studies investigating the role of TNF-a in neurology
The association of TNF with cardiovascular diseases
The pathogenesis of cardiovascular diseases involves many conventional risk factors, such as dyslipidemia, hypertension, obesity, diabetes, and smoking, as well as emerging risk factors, including genetics factors. Studies have shown that inflammation plays a role in the pathophysiology of atherosclerosis and CAD and is increasingly seen as a new modifiable cardiovascular risk factor. Inflammatory markers are important mediators in the multi-step cascade of atherosclerosis, which finally leads to the rupture of the atherosclerotic plaque [85].
Coronary artery disease
In Western societies, coronary artery disease (CAD) is associated with high morbidity and mortality and has therefore become a major public health challenge [86]. The pathogenesis of atherosclerosis is multifactorial and diverse. It is associated with the activation of several signaling pathways through inflammatory or mechanical damage to the vascular endothelium, thus mobilizing the body’s immune processes and involving pro-inflammatory cytokines [87, 88].
Furthermore, there is increasing recognition of inflammation as a novel modifiable risk factor for cardiovascular disease. TNF is a well-established pro-atherosclerotic factor [89]. Binding of TNF to its receptor (TNF-R1 or TNF-R2) on the surface of the cell membrane leads to the activation of cytokine production and initiation of the arachidonic acid cascade. This increases the intracellular concentration of free radicals and stimulates the liver to produce acute phase proteins and adipose tissue to produce adiponectin [90]. As a pro-inflammatory cytokine, TNF can be produced by cardiomyocytes. This occurs under unfavorable hemodynamic conditions, excessive pressure, or volume load. Consequently, it can lead to myocardial metabolic dysfunction, reduced peripheral blood flow, left ventricular dysfunction or its reconstruction [91]. The precise mechanism through which TNF exerts this effect remains unclear. An inverse correlation was observed between TNF and the E/A ratio [92]. In contrast, in patients with atrial fibrillation, the level of TNF correlates with the volume of the left atrium, which is a reliable indicator of diastolic function. Higher plasma TNF concentrations are associated with a higher risk of left ventricular hypertrophy and therefore may be a potential risk factor for diastolic dysfunction in patients with early CAD [93, 94]. TNF levels are also inversely correlated with ejection fraction [95] and QT prolongation, even in patients without heart failure. TNF is responsible for this phenomenon through the stimulation of reactive oxygen species (ROS). The “electric storm” of recurrent ventricular arrhythmias observed in heart diseases is also associated with elevated levels of this cytokine [96]. TNF levels measured 48 h after a cardiovascular episode have been shown to have a sensitivity of 78% and a specificity of 72.5% in predicting the occurrence of ischemic cardiovascular incidents, including angina, re-infarction, heart failure, and death [97, 98]. It has been demonstrated that the likelihood of death can be predicted by the degree of TNF elevation, which reduces myocardial contractility in patients with heart failure by disrupting intracellular calcium homeostasis [99]. Studies have also shown an association between elevated levels of inflammatory markers, including TNF-α, with the severity of CAD and the likelihood of future cardiovascular occurrences. This phenomenon can be attributed to the adaptive immune response of the body, which is caused by the prolonged retention of plasma lipoproteins and subsequent oxidative modification. This ultimately leads to endothelial damage. Recruited immune cells coordinate early atherosclerotic lesions by releasing pro-inflammatory cytokines, thereby accelerating foam cell formation, and ultimately promoting coronary syndrome through various inflammatory cascades [100]. Pericardial adipose tissue represents a small yet distinctive depot of fat, situated in close proximity to the cardiac muscle and coronary arteries. This proximity allows for the penetration of cells and proteins into the surrounding tissues [101]. TNF-α, derived from epicardial adipocytes, is the major mediator of local inflammation in several areas surrounding coronary arteries in patients with advanced CAD [102]. Coronary artery bypass graft (CABG) surgery, which is used to save the life of people CAD, can also induce an inflammatory response, release cytokines, including TNF, and lead to complications [103]. It is established that mean TNF levels in blood serum are significantly higher in patients with arterial hypertension and patients with acute myocardial infarction or stroke [104, 105]. Increases in circulating pro-inflammatory cytokines in patients with heart failure, particularly TNF, may reduce NO (nitric oxide) synthesis by reducing the expression of endothelial synthase of nitric oxide – a key enzyme involved in NO production. NO, however, acts as a regulator of muscle tension in the vascular endothelium/muscularis mucosae system. It is also an inhibitor of adhesion, platelet aggregation, and angiogenesis [106]. TNF promotes hypercholesterolemia and hypertriglyceridemia by reducing the lipoprotein lipase activity in adipocytes, prolonging low-density lipoprotein (LDL) retention in vascular walls and activating NF-κB and peroxisome proliferator-activated receptor γ (PPAR-γ) [107, 108]. The atherosclerotic effect of TNF inhibits high-density lipoprotein (HDL) and reduces cytokine release [109, 110]. In general, inverse associations have been observed between ApoA1, HDL-C, and ApoA1/HDL-C ratios and levels of inflammatory markers, including TNF-α. Furthermore, statins have been demonstrated to reduce TNF levels in plasma [111–113]. This is of particular importance in the case of patients with coronary artery disease. An inverse relationship between HDL and TNF-α levels has also been observed in patients with cerebrovascular ischemia [114, 115].
There is a dearth of publications documenting the strong statistical relationship between cytokines and measurements of atherosclerotic vascular lesions in Doppler studies. It is established that the concentration of soluble TNF receptors in serum is significantly correlated with the thickness of atherosclerotic plaques [116–118] and that the expression of TNF receptors on monocytes is positively related to the thickness of the inner and middle carotid artery [119]. In elderly patients, the length, thickness, and number of carotid atherosclerotic plaques, as well as TNF levels in plasma, decrease after they are treated with statins [120]. In contrast, in cultured endothelial cells, the neuropeptide catestatin (an endogenous nicotine cholinergic antagonist) inhibited the TNF-α-induced expression of adhesion molecules and inflammatory cytokines by activating angiotensin-2 converting enzyme (ACE2). Administration of the drug reduces interaction of leukocytes with the endothelium and acts as a pleiotropic cardioprotective hormone [121].
Inflammation plays an important role in atherosclerosis and CAD. Cytokines produced by immune cells, such as TNF-α, can be the target of therapeutic and anti-inflammatory molecules. The main anti-inflammatory therapies are aspirin, statins, colchicine, IL-1β, and IL-6 inhibitors, and new therapies such as TNF-α inhibitors and IL-1 receptor antagonists. Aspirin and statins reduce TNF levels, but TNF inhibitors are not yet used in clinical practice. The effects of TNF-α inhibitors such as infliximab, etanercept, and adalimumab on atherosclerosis have been investigated in preclinical studies. Infliximab reversed TNF-α-induced endothelial dysfunction. Etanercept may prevent TNF-induced endothelial cell apoptosis, while adalimumab may improve endothelial function and inflammation in patients. A meta-analysis of epidemiological and randomized control trials found that patients on anti-TNF therapy had a reduced risk of cardiovascular events, but this effect was not significant [100, 122–127].
CAD is a multifactorial disease in which many predictors are known, but new research is emerging on their dependence on signaling pathways. The introduction of new drugs, such as TNF inhibitors, must therefore consider the complexity of metabolic interactions [128]. Increased PCSK9 expression in the liver is positively associated with TNF-α, but a recent study reported no significant effects on leukocytes, monocytes, or neutrophils and no significant reduction in TNF-α after treatment with PCSK9 antagonists [129]. Different classes of drugs commonly used in the treatment of CAD have been shown to have different effects on inflammatory biomarkers. However, the current knowledge on inflammation and cardiovascular risk points to the importance of diagnostic criteria and evaluation of the anti-inflammatory and pro-inflammatory profile of the main treatment used in CAD therapy for clinicians [123, 130]. Knowledge of the inflammatory basis of CAD has not yet been supported by several large, controlled intervention trials. Selective TNF inhibitors may have led to unknown complications. A follow-up study is needed to improve their safety. A further trial investigating the efficacy of anti-inflammatory treatment in CAD patients needs to evaluate the effects of low-dose treatment versus placebo and assess the effects on the lipid profile.
Stroke
The redox reaction and stress are etiologies of vascular lesions [131]. The major reactive oxygen species (ROS) produced in response to several stimuli, such as hyperglycemia, hyperlipidemia, and hypertension, is superoxide anion (O2•−). It combines with NO to produce peroxynitrite, leading to endothelial dysfunction by reducing NO bioavailability. In response, endothelial cells are activated and produce vasoconstrictor agents: endothelin-1, thromboxane A2, or prostaglandin H2. Thus, an inflammatory response is initiated. The endothelium starts expressing adhesion molecules and secretes chemokines to attract immune cells. The increased expression of chemokines and proteases in endothelial cells perpetuates the inflammatory response [132]. In stroke, the lack of oxygen and nutrients can cause neurovascular units to become dysfunctional [133], affecting the production of ROS and the secretion of pro-inflammatory mediators [134]. Blood vessel occlusion or underperfusion affects the immune response near the ischemic parenchyma and then extends to the ischemic zone. Microglia are activated and promote the release of TNF-α [135]. TNF levels in brain tissue increase up to one day after ischemic injury and correlate with its severity [136]. The effects of TNF on the vascular endothelium include: oxidative stress through stimulation of xanthine oxidase expression, expression of tissue factors, production of leukocyte adhesion molecules, and activation of matrix metalloproteinases. These actions trigger local vascular inflammation, bleeding, and thrombosis [137]. TNF-α also disrupts the protective barrier between the brain and blood circulation, affects the activation and proliferation of astrocytes and microglia, and regulates apoptosis factors such as cysteine secretion and the transcription of metalloproteinases and inflammatory cytokines [138]. TNF-α has neurotoxic or neuroprotective effects in stroke [139]. In several studies, TNF-α has been associated with movement disorders, aphasia, epileptic seizures, pain, and cognitive impairment [140–142]. TNF is used as a potential therapeutic target in stroke, and its blockade reduces focal ischemic injury and improves clinical outcomes [143]. Therefore, detection of high levels of TNF may be useful as an inflammatory marker to predict stroke outcome. There are three effective ways to interfere with TNF: blocking TNF receptors, interfering with TNF signaling, and removing TNF protein [144]. TNF-α inhibitors – including infliximab, etanercept, adalimumab, golimumab, and pertuzumab – are currently used to treat inflammatory or autoimmune diseases, but may also reduce the risk of stroke [145]. The safety of anti-TNF agents and their lower efficacy in penetrating the blood-brain barrier need further investigation [146]. The role of TNF-α in cardiovascular diseases is summarised in Table III.
Table III
Summary of selected studies investigating the role of TNF-a in cardiovascular diseases
Conclusions
In response to harmful stimuli or dysregulation in the CNS and cardiovascular system, pro-inflammatory cytokines, such as TNF-α, are released by endothelial cells, neurons, and glial and infiltrating immune cells. The inflammation may be either harmful or protective, depending on various factors, including the initiating stimuli, targets, transcriptional changes, and the resulting intercellular activities. For this reason, overexpression of TNF-α should be considered an effect of deleterious stimuli. Elevated levels of this pro-inflammatory cytokine may cause neuronal excitotoxicity, synapse loss, and the development of neurological and cardiological diseases. However, the importance of TNF-α as a biomarker is ambiguous. For instance, although some current studies do not support TNF-α as a clear marker of stroke, it seems to be a promising research direction to use TNF-α as a biomarker of the stroke development process or prognosis [142]. The average cost of a TNF assay in the laboratory is around 30 euros. The recognition of TNF-α as a potentially valuable biomarker in neurodegenerative diseases is increasing. However, monitoring its levels and applying anti-TNF-α medications have not yet demonstrated significant clinical relevance. Another interesting aspect of TNF research is the importance of this pro-inflammatory cytokine in the aging process. Lindbergh et al. reported that plasma TNF-α concentrations were significantly associated with changes in white matter hyperintensities (WMH), gray matter volume (GMV), and global cognition. The authors suggested that systemic inflammation, as indexed by plasma TNF-α, holds potential as a biomarker for age-related declines in brain health [147]. Inflammation is increasingly recognized as a new modifiable cardiovascular risk factor. As a pro-inflammatory cytokine, TNF can be produced by cardiomyocytes, which typically occurs under unfavorable hemodynamic conditions with excessive pressure or volume overload. It can lead to myocardial metabolic disorders, left ventricular dysfunction, reduced peripheral flow, and left ventricular remodeling. The association of CAD with subclinical inflammation, quantified by values of circulating markers associated with inflammation, remains debatable. There are data supporting associations between circulating hs-CRP and cardiovascular outcomes. However, there is insufficient evidence showing that treatments to target inflammation and TNF levels result in improvements in patients. Therefore, plasma TNF levels are not a reliable marker of vascular status to replace the classic radiological examination of the vessels [94, 118, 148].
In conclusion, the basis for assessing the importance of TNF-α signaling in diseases of affluence, including neurodegenerative and cardiovascular diseases, is an understanding of the complex nature of the cytokine in question, as well as the involvement of TNF-α in regulating the release of other factors involved in physiological, inflammatory, or autoimmune processes. As our understanding of TNF-α deepens, we are closer to developing innovative prevention and treatment strategies that take genetic predisposition into account. It is a promising direction of research to use TNF as a biomarker for the development or prognosis of the described diseases. The conclusions need verification by clinical trials. To make TNF a reliable marker, the specific role and mechanism it plays, the protective effect and mechanism of anti-TNF-α treatment, and how to reduce the side effects of antibodies are the main issues that need to be further studied and solved by researchers. On the other hand, anti-TNF therapy is also worth exploring as a therapeutic target. The identification of intrinsic cell survival pathways should provide more direct opportunities for trials in neuroprotection or cardiovascular disease.