Introduction
Dementia is a progressive syndrome characterized by cognitive decline beyond normal aging, affecting memory, thinking, and daily functioning [1, 2]. It poses a significant global health challenge, particularly in aging populations globally [3]. As the global population continues to age, the current global estimate of 50 million individuals with dementia is projected to triple by 2050, reaching 150 million, necessitating innovative approaches to address this burgeoning public health crisis [4]. Despite its impact, public awareness remains low, and research is limited [3]. Current pharmacological interventions provide only symptomatic relief, yet they are often accompanied by adverse effects that may pose significant challenges in clinical management, such as nausea, vomiting, insomnia and even seizures [5]. This highlights the urgent need for new diagnostic techniques and therapeutic strategies with fewer side effects. Non-pharmacological therapies are increasingly important in delaying or preventing unhealthy aging [1].
Obstructive sleep apnea (OSA) is a common respiratory disorder characterized by recurrent upper airway collapse during sleep, leading to intermittent hypoxemia and sleep fragmentation [6]. Its prevalence ranges from 2% to 10% worldwide, with higher rates in middle-aged men (14–49%) [7, 8]. Risk factors include obesity, age, male gender, and smoking [8, 9]. OSA significantly impacts quality of life and is associated with increased risks of hypertension, cardiovascular diseases, stroke, diabetes, and heart failure [10–12]. Given its prevalence and health consequences, early recognition and treatment of OSA are key to preventing adverse outcomes.
Recent observational studies have increasingly highlighted a strong link between OSA and cognitive decline, with OSA being associated with an elevated risk of developing dementia, including Alzheimer’s disease (AD) and vascular dementia [13, 14]. Specifically, OSA has been associated with alterations in brain morphology, demonstrable deficits in memory, attention, and executive control, as well as higher risks of developing AD and even Parkinson’s disease [14]. The prevalence of OSA is significantly higher in AD patients compared to cognitively healthy individuals [15]. Studies have investigated AD biomarkers in OSA patients, including cerebrospinal fluid, blood, neuroimaging, and nuclear medicine biomarkers. While continuous positive airway pressure (CPAP) treatment may alleviate some damage, it does not fully reverse it [16]. However, while observational studies have provided numerous clues regarding the association between sleep apnea and dementia, their findings are susceptible to various confounding factors and cannot definitively exclude the possibility of reverse causation. Therefore, elucidating whether a true causal relationship exists between sleep apnea and dementia, and determining the direction of this causality, are of critical clinical and scientific importance for developing effective dementia prevention strategies and improving cognitive outcomes in patients with sleep apnea.
Mendelian randomization (MR) is an epidemiological method that uses genetic variants, randomly allocated during meiosis, as instrumental variables (IVs) to assess the causal effect of an exposure on an outcome [17]. To be more specific, MR employs genetic variants that influence the exposure (susceptibility to sleep apnea) as proxies to test whether the exposure has a causal impact on the outcome (dementia). Under the core assumptions that these genetic variants are strongly associated with the exposure, independent of confounding factors, and affect the outcome solely through the exposure, MR can mimic the design of a randomized controlled trial. As genetic variants are determined at conception and are generally not influenced by postnatal environmental factors or disease status, MR analysis can substantially mitigate confounding bias and reverse causation, which often plague traditional observational studies, thereby providing more robust evidence for causal inference [18]. In recent years, MR studies have begun to explore causal relationships between various exposures and either dementia or sleep apnea; however, systematic investigations into the bidirectional causal links between sleep apnea (specifically differentiating OSA and generalized SA) and multiple dementia subtypes remain scarce.
Given this context, the present study aimed to use publicly available summary-level data from large-scale genome-wide association studies (GWAS) within a two-sample MR framework. We systematically investigated the potential bidirectional causal associations between sleep apnea and various types of dementia. We expected to unravel the complex causal network between these two common and impactful diseases, thereby providing novel genetic evidence to inform early dementia prevention and the long-term management of patients with sleep apnea, potentially identifying new avenues for intervention.
Material and methods
Study design
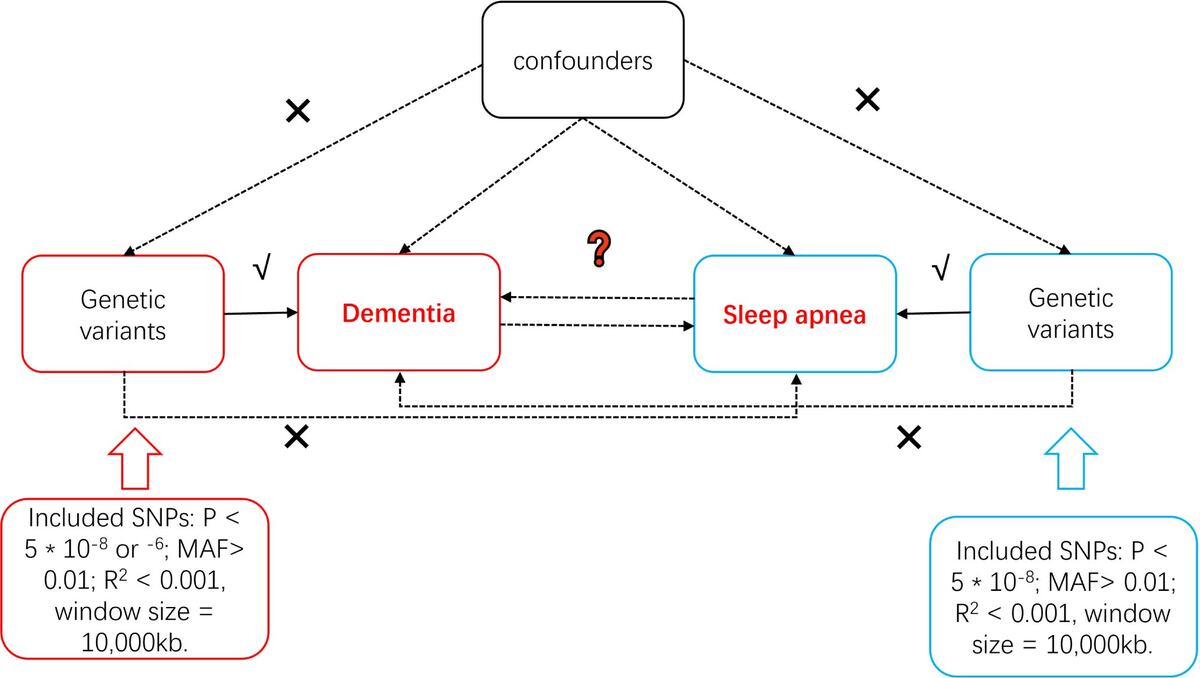
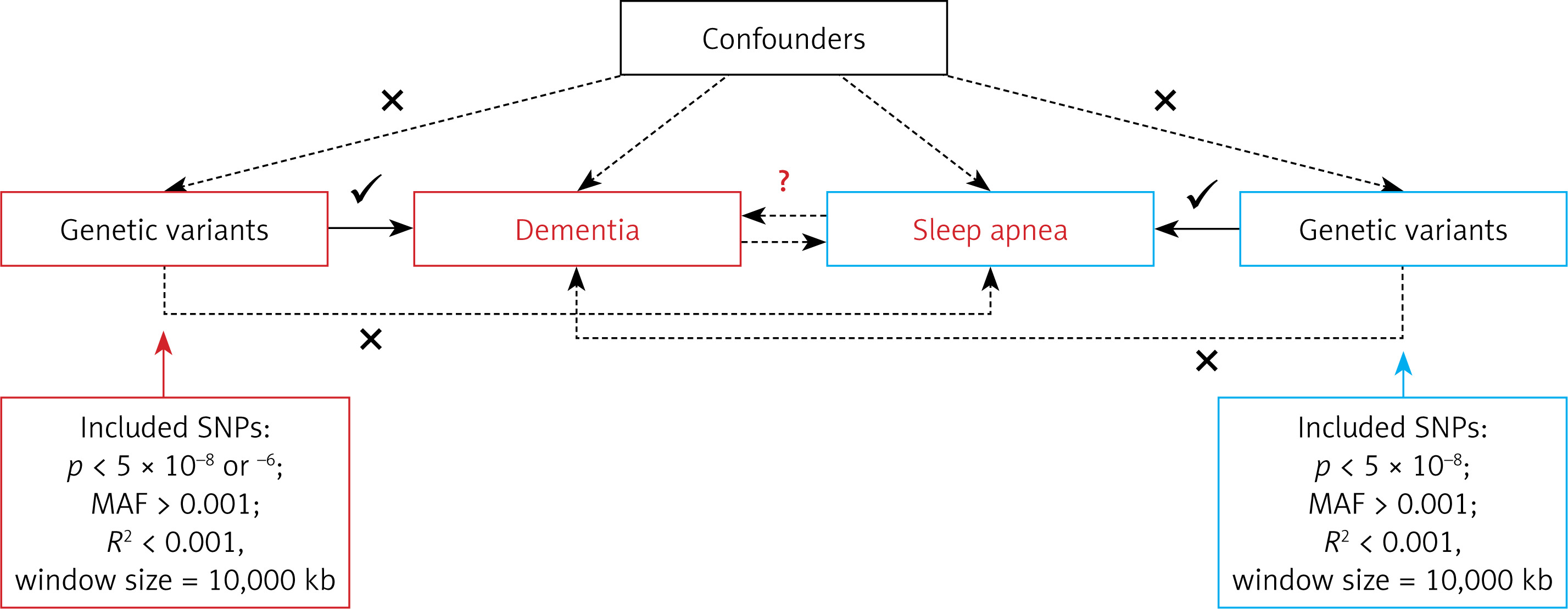
This study employed a two-sample MR study design. This methodology relies on several assumptions, as presented in Figure 1. For causal estimates derived from MR studies to hold validity, three fundamental assumptions must be satisfied: (1) IVs are robustly linked with the exposure; (2) IVs exhibit no association with potential confounding factors; (3) IVs do not directly influence the outcome, except through the exposure [19]. As this study is based on publicly available GWAS summary data, no further ethical approval was required. Our study is reported in accordance with the Strengthening the Reporting of Observational Studies in Epidemiology using MR (STROBE-MR) checklist [20].
Data sources
GWAS summary statistics for sleep apnea (including OSA and generalized sleep apnea) were sourced from a large published GWAS [21], primarily based on individuals of European ancestry. This included 126,695 cases and 303,630 controls for OSA, and 152,031 cases and 278,027 controls for generalized sleep apnea. GWAS summary statistics for various types of dementia were sourced from the FinnGen consortium (Release R12), which predominantly includes individuals of Finnish ancestry. Specifically, the “dementia” phenotype (GWAS ID: F5_DEMENTIA), comprising 24,864 cases and 469,981 controls, was defined based on International Classification of Diseases 10th Revision (ICD-10) codes F00-F09 from hospital discharge records. This block (F00-F09) comprises a range of mental disorders with a demonstrable etiology in cerebral disease, brain injury, or other insult leading to cerebral dysfunction. Other specific dementia phenotypes (e.g., unspecified dementia, vascular dementia, with sample sizes detailed in Supplementary Table SI) were also derived from FinnGen R12, based on their respective ICD-10 classifications. Both forward MR (sleep apnea as exposure, dementia as outcome) and reverse MR (dementia as exposure, sleep apnea as outcome) analyses were conducted. Detailed descriptions of the specific GWAS summary samples used in this study are presented in Supplementary Table SI.
Instrumental variable selection
To ensure the accuracy and reliability of conclusions regarding causal relationships, a series of quality control measures were implemented to select the most appropriate genetic instruments. Initially, SNPs that met the genome-wide significance threshold (p < 5 × 10–8) were used as IVs for sleep apnea exposures, and for dementia, unspecified dementia, and dementia in Alzheimer disease when these were considered exposures in the reverse MR [22]. For other dementia types as exposures, a threshold of p < 5 × 10–6 was applied. Only SNPs with a minor allele frequency (MAF) greater than 0.01 were retained. To minimize the impact of linkage disequilibrium (LD), SNPs were further filtered based on an LD threshold (r2 < 0.001) and a genomic region size limit of 10,000 kb [23]. The F-statistic, calculated as R2 × (N – 2)/(1 – R2) for each IV, was used to assess the instrumental strength, where R2 is the proportion of phenotypic variance explained by each genetic variant in the exposure, and N is the sample size [24]. An F-statistic exceeding 10 was set as the threshold for robust IVs [25]. In instances where selected SNPs were absent in the outcome summary data, proxy SNPs with high LD association (r2 > 0.8) were used as substitutes [26].
MR analyses
We conducted two-sample MR analyses using the TwoSampleMR package in R (version 4.0.5). The methodologies employed included inverse variance weighted (IVW), MR-Egger regression, weighted median (WM), and weighted mode approaches. Our principal analysis was based on the IVW method [25]. MR-Egger assesses causality while allowing for the presence of directional pleiotropy [27]. Weighted median analysis was used to assess causality, assuming validity in at least half of the IVs [19]. Initially, causal estimates were computed using the fixed effects IVW method for each IV. Upon detection of significant heterogeneity (p < 0.05), the analysis was shifted to the random effects IVW method. To visualize the causal relationships and the influence of each SNP on the outcome, forest, scatter, and funnel plots were generated. Multiple testing correction was applied using the false discovery rate (FDR) method, with associations deemed statistically significant at PFDR < 0.05.
Sensitivity and pleiotropy analysis
Pleiotropy detection was conducted via MR-Egger, where a significant intercept (p < 0.05) is indicative of pleiotropy [27]. Cochran’s Q test was used to evaluate heterogeneity among IVs for each phenotypic exposure, with a p-value above 0.05 signifying an absence of heterogeneity [28]. MR-PRESSO was employed to identify and eliminate outliers (p < 0.05) and to adjust for horizontal pleiotropy [29]. A leave-one-out analysis was performed to determine the influence of each SNP on the overall causality by excluding each SNP sequentially before conducting MR analyses on the remaining SNPs [30].
Results
Selection of IVs
In the forward analysis, when OSA and sleep apnea were exposures, we identified 78 and 112 IVs, respectively. The mean F-statistics for these IVs were 42.1 (OSA) and 40.41 (sleep apnea); minimum F-statistics were 29.75 (OSA) and 29.78 (sleep apnea); and maximum F-statistics were 380.34 (OSA) and 392.23 (sleep apnea), respectively. For 11 single nucleotide polymorphisms (SNP) not directly available in outcome datasets, proxy SNPs were used where possible (e.g., rs370508348 by rs7594555). An additional 4 SNPs were removed during data harmonization. Detailed data for IV selection in the forward analysis are provided in Supplementary Tables SII–SIV.
In the reverse analysis, the exposures included dementia due to Parkinson’s disease, dementia in Alzheimer disease, dementia in other diseases classified elsewhere, dementia (general), frontotemporal dementia, unspecified dementia, and vascular dementia, with the number of IVs being 13, 17, 20, 31, 2, 8, and 18, respectively. For the 13 IVs for dementia due to Parkinson’s disease, the mean F-statistic was 24.14, the minimum F-statistic was 21.14, the maximum F-statistic was 35.66, and the R2 value was 0.001442. For the remaining exposures, mean F-statistics ranged from 24.93 (dementia in other diseases classified elsewhere) to 220.72 (dementia in Alzheimer disease). All minimum F-statistics exceeded 20 (thus satisfying the F > 10 criterion), with the highest maximum F-statistic being 2628.18 (dementia in Alzheimer disease). When OSA or sleep apnea served as the outcome, 9 SNPs were unmatched in their summary datasets; proxy SNPs were employed where possible (e.g., rs140887881 by rs145562973). Subsequently, 2 SNPs were excluded for the OSA outcome and 5 SNPs for the sleep apnea outcome during harmonization. Detailed data for IV selection in the reverse analysis are provided in Supplementary Tables SII, SV, and SVI.
Mendelian randomization analysis
Forward analysis
In the initial forward analysis for the association between OSA and unspecified dementia, the IVW method did not show a statistically significant association (OR = 0.600, 95% CI = 0.31–1.160, p = 0.1275, Table I, Supplementary Figure S1). However, the weighted median method showed a significant protective effect (OR = 0.750, 95% CI = 0.590–0.950, p = 0.0178, Table I, Supplementary Figure S1). The MR-PRESSO analysis identified the presence of outliers for this association (Table II). After manual removal of outliers, the IVW analysis revealed a statistically significant association, indicating that OSA was associated with a reduced risk of unspecified dementia (OR = 0.830, 95% CI = 0.700–0.970, p = 0.021, FDR = 0.021, Table I, Supplementary Figure S2). This finding was further supported by the weighted median method (OR = 0.740, 95% CI = 0.580–0.940, p = 0.015, FDR = 0.015, Supplementary Figure S2) using the outlier-corrected dataset (Table I). Detailed data for all forward MR analyses, including results before and after outlier removal for all methods, are provided in Supplementary Table SVII, SVIII.
Table I
Causal associations
Table II
Heterogeneity tests and pleiotropy tests for instrumental variables
The MR-Egger method results showed no significant pleiotropy for the association between OSA and unspecified dementia after outlier removal (p for intercept = 0.767, Table III). For the IVW analysis of OSA and unspecified dementia after outlier removal, the Q statistic did not suggest significant heterogeneity (p = 0.535, Table III). Detailed sensitivity analysis data are provided in Supplementary Tables SIX–SXII.
Table III
MR-PRESSO results
Reverse analysis
Reverse analysis initially revealed several significant associations. General dementia was associated with a reduced risk of OSA (IVW method: OR = 0.9418, 95% CI = 0.9107–0.9740, p = 0.0005, FDR = 0.0017, Table I, Supplementary Figure S3) and generalized sleep apnea (IVW method: OR = 0.9412, 95% CI = 0.9122–0.9711, p = 0.0001, FDR = 0.0007, Table I, Supplementary Figure S4). The MR-PRESSO analysis detected outliers for both associations (Table II). After manual removal of these outliers, general dementia remained significantly associated with a reduced risk of OSA (IVW method: OR = 0.9399, 95% CI = 0.9187–0.9615, p < 0.001, FDR < 0.001, Table I, Supplementary Figure S5) and generalized sleep apnea (IVW method: OR = 0.9141, 95% CI = 0.8863–0.9427, p < 0.001, FDR < 0.001, Table I, Supplementary Figure S6).
Furthermore, unspecified dementia (IVW method: OR = 0.9644, 95% CI = 0.9334–0.9964, p = 0.0293, Table I, Supplementary Figure S7), dementia due to Parkinson’s disease (IVW method: OR = 0.9760, 95% CI = 0.9546–0.9979, p = 0.0315, Table I, Supplementary Figure S8), and dementia in Alzheimer’s disease (IVW method: OR = 0.9668, 95% CI = 0.9514–0.9824, p < 0.001, FDR = 0.0003, Table I, Supplementary Figure S9) were associated with a reduced risk of OSA. Dementia in Alzheimer’s disease was also associated with a reduced risk of generalized sleep apnea (IVW method: OR = 0.9680, 95% CI = 0.9552–0.9809, p < 0.001, FDR < 0.001, Table I, Supplementary Figure S10).
Furthermore, vascular dementia was associated with a reduced risk of sleep apnea (IVW method: OR = 0.9671, 95% CI = 0.9419-0.9931, p = 0.0133 FDR = 0.031, Table I) and OSA (IVW method: OR = 0.9668, 95% CI = 0.9596–0.9845, p = 0.0016, FDR = 0.0044, Table I). However, MR-PRESSO analysis identified outliers for this association, and after their manual removal, the association was no longer statistically significant. Detailed data for all reverse MR analyses, including results before and after outlier removal where applicable, and their corresponding FDR values, are provided in Supplementary Tables SXIII, SXIV.
For the reverse causal associations that remained significant after outlier removal, such as general dementia with OSA (Cochran’s Q p = 0.154; MR-Egger intercept p = 0.343) and generalized sleep apnea (Cochran’s Q p = 0.218; MR-Egger intercept p = 0.701), sensitivity analyses indicated no significant heterogeneity and horizontal pleiotropy (Table III). Other positive reverse associations (unspecified dementia with OSA, dementia in Alzheimer’s disease with OSA, and dementia due to Parkinson’s disease with OSA) also showed no evidence of pleiotropy via the MR-Egger intercept, although some exhibited heterogeneity (Table III). This observed heterogeneity is generally permissible given that the primary IVW method employed in this study used a random-effects model. Detailed sensitivity analysis data, including results for associations before outlier removal, are provided in Supplementary Tables SXV–SXVIII.
Discussion
This study employed a two-sample MR approach to systematically investigate potential causal relationships between sleep apnea and various dementia subtypes. Our principal findings suggest a causal protective effect of genetic predisposition to OSA on the risk of unspecified dementia. Intriguingly, our analyses also suggested potential causal associations between genetic predisposition to general dementia and dementia in AD and a reduced risk of both OSA and generalized sleep apnea, although these findings warrant cautious interpretation. These results offer novel genetic perspectives on the complex interplay between sleep apnea and dementia, holding potential implications for disease prevention and management.
The observed causal association suggesting that OSA may reduce the risk of unspecified dementia (IVW OR = 0.830) is a noteworthy phenomenon that warrants careful consideration. This finding appears to contradict a substantial body of observational research reporting OSA as an independent risk factor for dementia [31–33]. This discrepancy may be attributable to the inherent strength of MR in mitigating confounding, a common limitation in observational studies. Alternatively, it could stem from the heterogeneity within the “unspecified dementia” outcome. This category is broadly defined to include various conditions such as “dementia NOS”, “psychosis NOS”, and “primary degenerative dementia NOS” (both presenile and senile), while excluding specific diagnoses such as “senile dementia with delirium” (F05.1) and “senility NOS” (R54). Consequently, “unspecified dementia” might encompass early-stage, atypical, or etiologically unclear cognitive impairments whose underlying pathophysiology could differ significantly from that of well-defined dementia subtypes.
One potential explanation is that the genetic predisposition to OSA identified by MR analysis might differ from the clinically diagnosed OSA phenotype with significant pathophysiological consequences. Individuals carrying certain OSA genetic risk alleles might experience mild, subclinical intermittent hypoxia exposure throughout their early life, particularly as many risk factors for OSA are genetically determined [34]. While chronic, severe intermittent hypoxia undoubtedly promotes neurodegeneration through oxidative stress, neuroinflammation, impaired cerebral autoregulation, and accelerated Aβ deposition [35], the existence of a “hypoxic preconditioning” or “mild stress adaptation” window is debatable [36]. Some studies suggest that mild intermittent hypoxia may induce neuroprotective preconditioning effects, mediated by hypoxia-inducible factor (HIF) and its downstream targets such as vascular endothelial growth factor (VEGF) and erythropoietin [37], thereby enhancing neuronal tolerance to subsequent, more severe insults and promoting neurovascular unit remodeling. If individuals genetically predisposed to OSA experience a lifelong pattern of hypoxic exposure that includes such preconditioning effects, possibly mediated by HIF and VEGF, and if this adaptive response outweighs the direct damage from mild hypoxia, it might confer a degree of “relative protection” against certain “vulnerable” or slowly progressing types of unspecified dementia. However, this remains a highly speculative explanation, requiring more refined mechanistic studies and MR analyses stratified by OSA severity. Furthermore, undetected horizontal pleiotropy of the IVs cannot be entirely excluded, meaning that these OSA-related genetic variants might influence the risk of unspecified dementia through pathways independent of OSA.
Regarding the unexpected MR finding that genetic liability to general dementia and dementia in AD might be causally associated with a reduced risk of sleep apnea, careful consideration is warranted. This genetically inferred directionality is counterintuitive, contrasting with clinical observations that often associate manifest dementia with increased sleep apnea prevalence due to factors such as pharyngeal muscle weakness or later-stage medication effects [38–40]. While speculative, one potential biological avenue for such a genetically driven protective effect could involve early, subtle dementia-related alterations in neurotransmitter systems (such as cholinergic pathways [41]) or sleep-wake regulation that paradoxically stabilize respiration before widespread neurodegeneration. However, this counterintuitive observation could also stem from other factors. Complex genetic pleiotropy, where dementia-associated variants independently influence pathways reducing sleep apnea risk, is a possibility. Methodological considerations, such as potential survival or selection biases in GWAS for late-onset diseases or residual confounding from uncharacterized horizontal pleiotropy, are also pertinent [29]. Furthermore, the lifelong effect of genetic liability captured by MR may not fully align with the multifaceted clinical picture of advanced dementia.
The careful selection of datasets for dementia in AD and PD in this study, emphasizing ICD codes distinct from isolated AD or PD, helps to more accurately capture the causal effect of the “dementia state” itself on sleep apnea. GWAS data for AD (ICD-10, G30) or PD (ICD-10, G20) might predominantly represent earlier disease stages, often without overt dementia or associated severe neuromuscular or central respiratory dysregulation. In contrast, analyses focusing on “dementia in AD (F5_ALZHDEMENT)” or “dementia in PD (PD_DEMENTIA_EX)” are more reflective of the impact when neurodegenerative processes have advanced sufficiently to cause a dementia syndrome.
The strengths of this study lie in its use of the MR design, effectively controlling for confounding inherent in observational studies and allowing for the assessment of potential causal directions. We used summary data from large-scale GWAS, enhancing statistical power. Furthermore, analyzing multiple dementia subtypes and differentiating between OSA and generalized sleep apnea provided more nuanced insights. The application of various sensitivity analyses also bolstered the robustness of our findings. However, this study also has limitations. First, MR analysis relies on three core assumptions; while we conducted several sensitivity analyses to assess these (e.g., for horizontal pleiotropy), the potential influence of unknown pleiotropy cannot be entirely ruled out. Second, the GWAS data used were primarily from European and Finnish populations, which may limit the generalizability of our findings to other ethnic groups. Third, despite distinguishing between dementia subtypes, GWAS data may not fully capture the heterogeneity within these conditions, such as dementia severity or disease duration. Fourth, for some dementia subtypes (e.g., frontotemporal dementia), the limited number of cases might have resulted in insufficient statistical power and weaker IVs, leading to uncertain results. Fifth, given that MR analyses are grounded in genetic susceptibility and diverge from clinical observational studies, it is plausible that sleep apnea treatment could modulate dementia risk in clinical settings, though this requires further validation in future clinical studies. Finally, for several of the dementia-to-sleep-apnea associations, the observed odds ratios, while reaching statistical significance in some analyses, were modest. Such effect sizes warrant cautious interpretation as they might represent smaller true effects or be more influenced by subtle biases, underscoring the need for validation in significantly larger samples.
Future research must prioritize replication of these findings, especially the unexpected association between dementia predisposition and reduced sleep apnea risk, in independent, large-scale, and diverse cohorts. Investigating potential methodological reasons for this counterintuitive finding should be a priority. Further studies should also aim to use more refined phenotypes and investigate potential mediating biological pathways if these causal associations are substantiated. In addition, clarifying dementia subtypes may facilitate earlier risk prediction and may promote the development of subtype-specific therapeutic or precision prevention strategies.
In conclusion, this MR study provides suggestive genetic evidence supporting a causal protective effect of OSA predisposition on unspecified dementia risk. It also presents exploratory, and more tentative, genetic evidence suggesting that predisposition to general dementia and AD may be causally associated with a reduced risk of sleep apnea. While the former aligns with hypotheses such as hypoxic preconditioning, the latter is unexpected and requires substantial further investigation to distinguish potential novel biological insights from methodological or complex genetic phenomena. These findings underscore the intricate, and potentially counterintuitive, causal dynamics between sleep apnea and dementia.