Introduction
Sepsis persists globally as a leading cause of mortality, responsible for nearly 11 million annual deaths worldwide [1]. Defined as life-threatening organ dysfunction induced by a dysregulated host response to infection [2], sepsis manifests clinically as an exuberant inflammatory response culminating in cardiovascular collapse and multiorgan failure [3]. Various circulating inflammatory mediators released during sepsis contribute to its pathogenesis, but their precise causal roles remain poorly defined [4]. Elucidating impacts of these molecules could provide vital insights into mechanisms of sepsis while highlighting proteins of interest for clinical translation [5].
Mendelian randomization (MR) utilizes genetic variants as instrumental variables to infer causal relationships free of biases inherent to traditional observational research [6–8]. By integrating publicly available genome-wide association study (GWAS) summary statistics, two-sample MR enables efficient large-scale evaluation of causal effects for diverse exposures [9, 10]. Here we performed a comprehensive two-sample MR study to investigate potential causal impacts of circulating inflammatory proteins on sepsis risk. Our findings help delineate roles of key inflammatory mediators in sepsis development, while providing targets for future mechanistic and clinical investigation.
By screening for cytokine genetic instruments and evaluating their associations with extensive sepsis outcomes, our approach facilitates hypothesis-free assessment of cytokine roles in sepsis pathogenesis. The findings help delineate immunological drivers of sepsis development and mortality, while providing putative prognostic biomarkers and therapeutic candidates worthy of further evaluation to improve sepsis management.
Material and methods
Research design
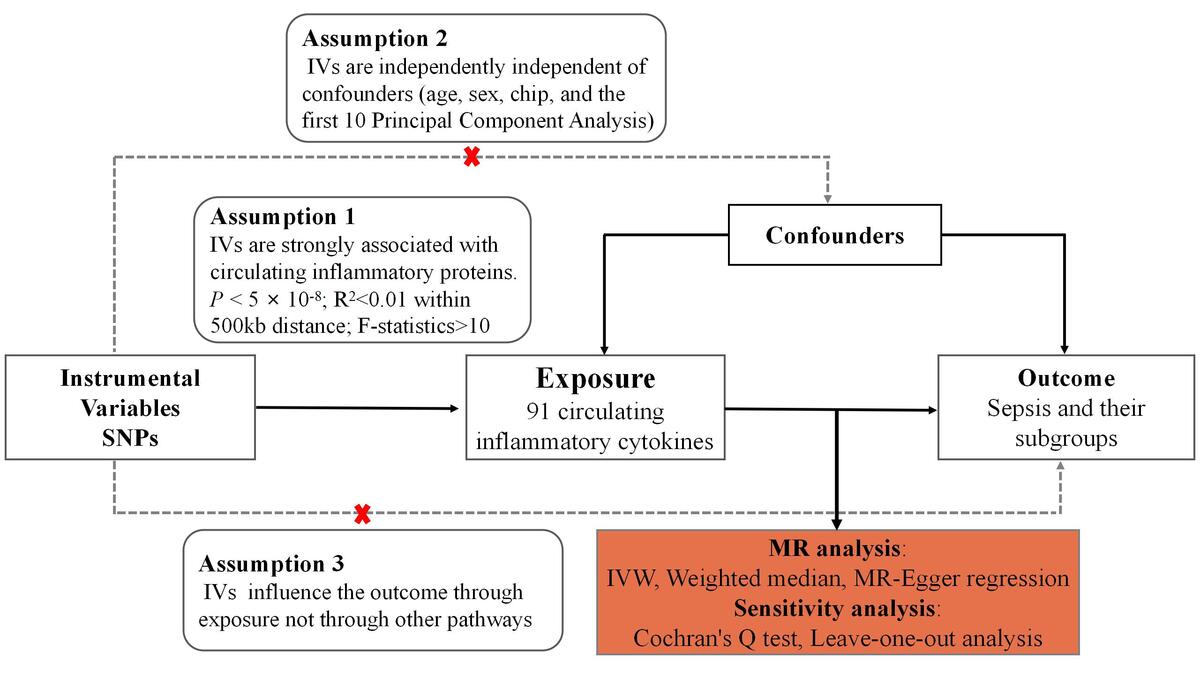
We performed an MR analysis based on publicly available pooled data from genome-wide GWAS analyses to assess the causal relationship between 91 circulating inflammatory proteins and sepsis. To minimize population stratification bias, the cohorts of circulating inflammatory proteins and sepsis were subjects of European ancestry. All data used in this study were publicly available from studies that received consent and ethical approval from the participants involved; therefore, this study did not require ethical approval from an institutional review board. This study followed the 2021 Strengthening Observational Research in Epidemiology Report (STROBE) and STROBE-MR guidelines [11]. MR analysis and sensitivity analysis were performed successively. In MR analysis, the following key assumptions were made in this study: (1) the IVs are strongly associated with circulating inflammatory proteins; (2) the IVs are independently independent of confounders; (3) the IVs affect the outcome only through circulating inflammatory proteins [12]. Figure 1 sums up the entire study design and workflow.
Data sources
Data related to circulating inflammatory proteins were publicly available from the GWAS catalog (GCST90274758 to GCST90274848) [3]. Zhao et al. conducted a genome-wide protein quantitative trait locus (pQTL) study of plasma proteins in 14,824 participants through the Olink Target platform, identifying proteins associated with 91 circulating inflammatory cytokines [13]. As for the sepsis cases and their subgroups (under 75, 28-day death, intensive care units [ICU], 28-day death in ICU), they were collected from the IEU Open GWAS with summary-level data obtained from the UK Biobank, which included 11,643; 11,568; 1,896; 1,380 and 347 sepsis cases and 474,841; 451,301; 484,588; 429,985; 431,018 controls, respectively [14]. The UK Biobank study is a large multicenter cohort study that recruited more than 500,000 European participants across the United Kingdom between 2006 and 2010 [15]. This study used Regenie v2.2.4 to analyze GWAS data, and adjusted for age, sex, chip, and the first 10 principal component analysis. In order to check the stability of our results, we replicated the results in additional data from the FinnGen consortium, including 12,301 cases and 227,388 controls. The detailed information is listed in Table I.
Screening of instrumental variables
In this study, we selected genetic IVs that met the following conditions: (1) the SNPs associated with each of the 91 cytokines reached the genome-wide significance threshold (p < 5 × 10–8); (2) to avoid linkage disequilibrium (LD) among the included IVs, an R2 test was performed to minimize bias with the clumping process of R2 < 0.01 and clumping distance = 500 kb; (3) to ensure a strong estimated effect of genetic variation, we included SNPs with F > 10 [16]. To test for the presence of bias in the IVs, – whether genetic variation in the IVs was weakly associated with cytokines – we calculated the F-statistic [F = R2 (n – 2)/(1 – R2)], where R2 is the variance of cytokines explained by IVs, and n is the sample size [17]. An F-statistic greater than 10 means that weak instrumental variable bias is less likely [18]. The main information of SNPs, including effect allele, other allele, beta, standard error, and p-value, was collected systematically for further analysis. Finally, we identified 558 SNPs for the circulating levels of 74 cytokines. After excluding 60 SNPs for which associations of these SNPs were not present in the summary-level data, we used the remaining SNPs associated with the circulating level of 72 cytokines as IVs in primary MR analyses.
Statistical analysis
We conducted the MR analysis with several methods to estimate the potential causal associations between circulating level of cytokines and sepsis, including the inverse-variance weighted (IVW) method, weighted median method, and MR-Egger regression. The IVW method was chosen as the primary analysis method to evaluate the relationships [19]. The IVW method is an effective analysis on the assumption that all genetic variations are effective instrumental variables and almost all MR analyses use it as the main analysis for its accuracy [19]. Thus, only when the processed data withstand all the sensitivity analyses can the result of IVW be considered sufficiently reliable. Therefore, we used Cochran’s Q test to test the heterogeneity of the IVs we selected [20]. If the heterogeneity exists (p < 0.05), the random-effects IVW method would be chosen or the weighted median method may be more credible [20, 21]. Second, MR-Egger regression was performed to assess the potential presence of the horizontal pleiotropy, with p-values for the intercept < 0.05 considered to indicate the existence of pleiotropy [22]. In the case of pleiotropy, an MR-Egger regression estimate might be preferred [22]. Finally, we applied the leave-one-out analysis to determine whether the removal of any IV could cause significant changes in the results.
We have presented the odds ratios (ORs) with their 95% confidence intervals (CIs) in the forest plots for the significant results (Figures 2, 3). Considering that our data were multiple comparisons based, stricter Bonferroni-corrected significance levels of p-values should be applied. For cytokines, we set the threshold at p < 1.35 × 10–4 (0.05/74/5). For the p-value of our main testing method, IVW, ranging from the Bonferroni-corrected significance level to 0.05, we considered this as evidence suggesting a potential causal association [13]. The two-sample Mendelian randomization analysis of our study was performed in the R software (v.4.2.3) with the R package TwoSampleMR (v.0.5.7).
Results
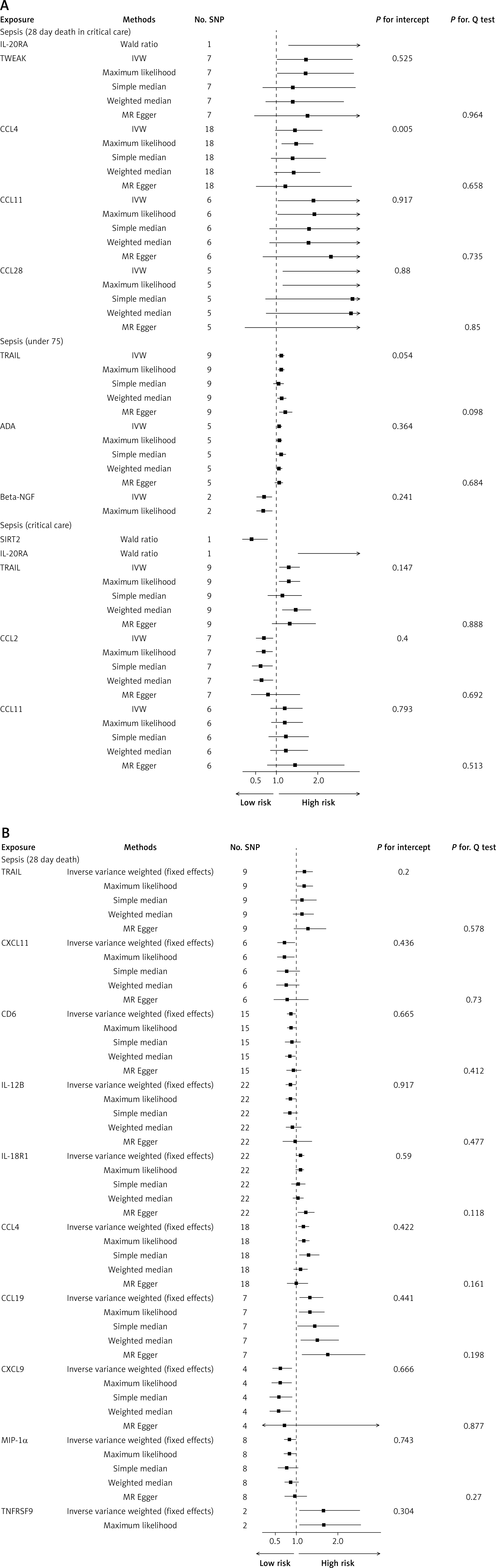
The F-statistics of IVs ranged from 29.72 to 1911.35, suggesting that the association between cytokines and sepsis would not be influenced by weak instruments (Supplementary Table SI). Among 72 cytokines in total, we found that three cytokines were significantly associated with sepsis (Figure 2). More details are presented in Figure 3. Beta-NGF and TNFB were negatively associated with sepsis (beta-NGF, OR = 0.77, 95% CI: 0.60–0.99, p = 0.039; TNFB, 0.95, 95% CI: 0.91–1.00, p = 0.031) in the IVW method, which was consistent with the maximum likelihood method. Also, there was a positive effect of TRAIL on sepsis (OR = 1.10, 95% CI: 1.03–1.17, p = 0.004), while it showed significant fluctuation after removing several SNPs.
Figure 3
The causality of Beta-NGF, TNFB and TRAIL with SS in the MR analysis. A - Beta-NGF; B - TNFB; C - TRAIL

Regarding sepsis sub-traits, we found that there were 18 cytokines that had causal relationships with them (Figure 2). Similarly, TRAIL level was positively associated with three sepsis traits: sepsis (critical care), sepsis (under 75), sepsis (28-day death). As shown in Figure 4, we found that CCL28 had a significant positive impact (OR = 3.32, 95% CI: 1.18–9.29, p = 0.022) on sepsis (28-day death in critical care). In addition, there was a protective effect of CCL2 on sepsis (critical care) in the IVW method, and it was similar in the other methods such as simple median, weighted median and maximum likelihood. This causality was consistent with the maximum likelihood method, and there was no heterogeneity or pleiotropy in Cochran’s Q test and MR-Egger (all p > 0.05). Among ten cytokines associated with sepsis (28-day death), CCL4 had the most significant association (OR = 1.81, 95% CI: 1.06–1.31, p = 0.002, Supplementary Table SV). The maximum likelihood and simple median showed a consistent association (Figure 4). The MR-Egger suggested that there was no influence of pleiotropy of IVs in the MR analysis. More details about the association between cytokines and sepsis are presented in Supplementary Tables SII–VI.
Discussion
In this comprehensive two-sample MR study, we identified multiple circulating inflammatory proteins demonstrating significant causal associations with sepsis risk. Our findings provide insight into immunological pathways underlying sepsis development while highlighting promising biomarkers and therapeutic candidates.
Our analyses identified TRAIL as an inflammatory cytokine exhibiting significant causal effects on increased overall sepsis risk as well as higher risk for sepsis-associated mortality. TRAIL emerged as the protein with the most extensive causal links to sepsis outcomes. This pro-inflammatory cytokine showed a causal effect on increased overall sepsis risk as well as elevated risk for sepsis in the young, sepsis mortality, and sepsis treated in critical care settings. TRAIL has been previously implicated as a mediator of immune cell apoptosis during sepsis leading to lymphopenia and impaired immunity [23, 24]. TRAIL has been previously shown to mediate immune cell apoptosis during sepsis leading to lymphopenia and impaired immunity, which aligns with our finding of its causal role in increasing sepsis risk. Our results provide strong evidence supporting a causal role of TRAIL in driving sepsis pathogenesis. Therapies targeting TRAIL signaling may therefore hold promise for future clinical translation.
The chemokine CCL28 also demonstrated a significant causal association in our study with increased mortality risk in sepsis patients treated in the ICU. Known functions of CCL28 include attracting T-cells and inducing mucosal healing responses [25]. The observed link of higher CCL28 levels with poor sepsis prognosis may reflect dysregulated chemokine activity contributing to mortality or attempted compensatory mechanisms. Additional research into mechanisms underlying this association is warranted.
We found the chemokine CCL4 to have the most significant causal relationship with overall 28-day mortality from sepsis. As a chemoattractant for immune cells such as monocytes and macrophages, aberrant CCL4 signaling can propagate harmful inflammation [26]. Our results designate CCL4 as a potential driver of the exaggerated inflammatory response leading to early sepsis death. CCL4 may also serve as a prognostic biomarker, although further longitudinal analyses are required.
Two inflammatory proteins emerged with potential protective effects against sepsis: the neurotrophins beta-NGF and TNFB. By signaling through tropomyosin receptor kinase A, beta-NGF regulates neuronal survival and inflammation [27]. Observational studies associate lower beta-NGF with worse sepsis outcomes, concordant with our findings [28]. Beta-NGF may therefore represent a protective pathway in sepsis that could be therapeutically augmented. TNFB is a tumor necrosis factor superfamily member modulating immune cell proliferation and death [29]. While TNF-α is a well-known sepsis mediator, less is known about TNFB’s role. Our results now identify TNFB as another protective factor in sepsis worthy of investigation.
Taken together, our study implicates circulating inflammatory mediators such as TRAIL, CCL28, and CCL4 as causal immunological drivers of sepsis risk and mortality, highlighting new therapeutic targets worthy of further investigation.
By integrating large GWAS datasets through MR methodology, our study enabled efficient hypothesis-free evaluation of causal links between circulating inflammatory proteins and sepsis risk. Utilization of genetic instruments as proxies for protein levels conferred advantages over traditional observational research by minimizing biases from confounding variables and reverse causation. We also employed multiple sensitivity analyses to confirm result validity and account for potential issues such as pleiotropy.
However, some limitations must be acknowledged. Despite the large samples, the power was modest for discovery of weaker effects. Ethnic generalization requires further assessment. Protein changes over time could not be examined owing to static measurements. The analyzed pathways likely represent only a fraction of the intricate immunological circuitry in sepsis, although they may be strategically poised as pharmacological targets upstream of cytokine cascades and cellular damage. While congruent with known sepsis biology, the identified proteins warrant direct functional validation. Lastly, we cannot definitively exclude pleiotropic effects of genetic instruments on unmeasured pathways that could influence the results. However, the consistent findings across MR methods with distinct assumptions mitigate this concern.
In conclusion, our MR study elucidates impacts of key inflammatory cytokines and chemokines on sepsis development. The identified circulating proteins likely contribute to sepsis outcomes through effects on pathways involved in immune cell apoptosis and exaggerated inflammation. By demonstrating causal relationships, these findings shed new light on immunological mechanisms underlying sepsis progression.