Introduction
Atherosclerotic cardiovascular disease (ASCVD), which encompasses two primary subtypes – coronary heart disease (CHD) and ischemic stroke (IS) – constitutes the foremost cause of mortality globally [1, 2]. Consequently, the associated global burden of ASCVD remains a critical public health priority on an international scale [3]. The development of ASCVD involves complex pathophysiologic processes involving genetic and environmental factors and their interactions over decades [4, 5]. Improved prevention and intervention strategies for ASCVD need to comprehensively summarize the impacts of environmental and genetic factors. The “developmental origins of health and disease” (DOHaD) hypothesis proposes that adverse developmental experiences early in life increase the risk of cardiovascular disease and metabolic comorbidities later in life [6–8]. During the vulnerable period of intrauterine development, disturbances in gene expression or cell proliferation and differentiation caused by nutritional and other environmental influences can alter the structure and function of life’s major organ systems, a process known as developmental programming. These effects set the stage for many diseases that manifest many years later, often in response to secondary environmental stressors [9].
The placenta, a temporary organ in mammalian gestation, comprises the chorionic villi, the placental mesenchyme, and the placental membrane. The chorionic villi represent the fetal component, whereas the placental mesenchyme constitutes the maternal component [10, 11]. At the end of pregnancy, the placenta is expelled by the mother. The primary functions of the placenta include providing nutrients and facilitating gas exchange, as well as secreting various hormones essential for maintaining a normal pregnancy. Furthermore, the placenta serves as a selective barrier, permitting the passage of nutrients and gases while obstructing potentially harmful substances from reaching the fetus [12]. Failure of the placenta to protect or nourish the fetus may have severe consequences for the fetus, both before birth and throughout its postnatal life [9, 13, 14]. Previous observational studies have demonstrated that placental health is associated with the risk of cardiovascular disease and metabolic comorbidities later in life [15–18]. Several animal experiments have also provided evidence that the placenta plays a vital role in regulating the development of the cardiovascular system [19–22].
Placental weight (PW) is easily measured and is considered an indicator for assessing placental growth and function. It is often used in epidemiologic studies to represent placental growth and function [15, 23, 24]. Previous research has indicated a correlation between PW and cardiovascular disease; however, the existence of a causal association remains uncertain [25–28]. A comprehensive investigation of the causal relationship between PW and cardiovascular disease, as well as the mediating factors involved in this association, could enhance our understanding of the etiological development of cardiovascular disease and offer valuable insights into its prevention and intervention strategies.
In this study, we employed the Mendelian randomization (MR) method to investigate the potential causal relationship and mediating factors between PW and ASCVD. MR methods apply genetic variation as an instrumental variable (IV) to infer causal relationships between related traits [29]. We used several extension methods of basic MR principles, including two-sample MR [30], two-step MR [31], and multivariate MR [32, 33], and then integrated the results into mediation analyses. For mediation analysis, the two-step MR strategy is sensitive to causal mediation effects and is less susceptible to measurement error [31].
Material and methods
Study design
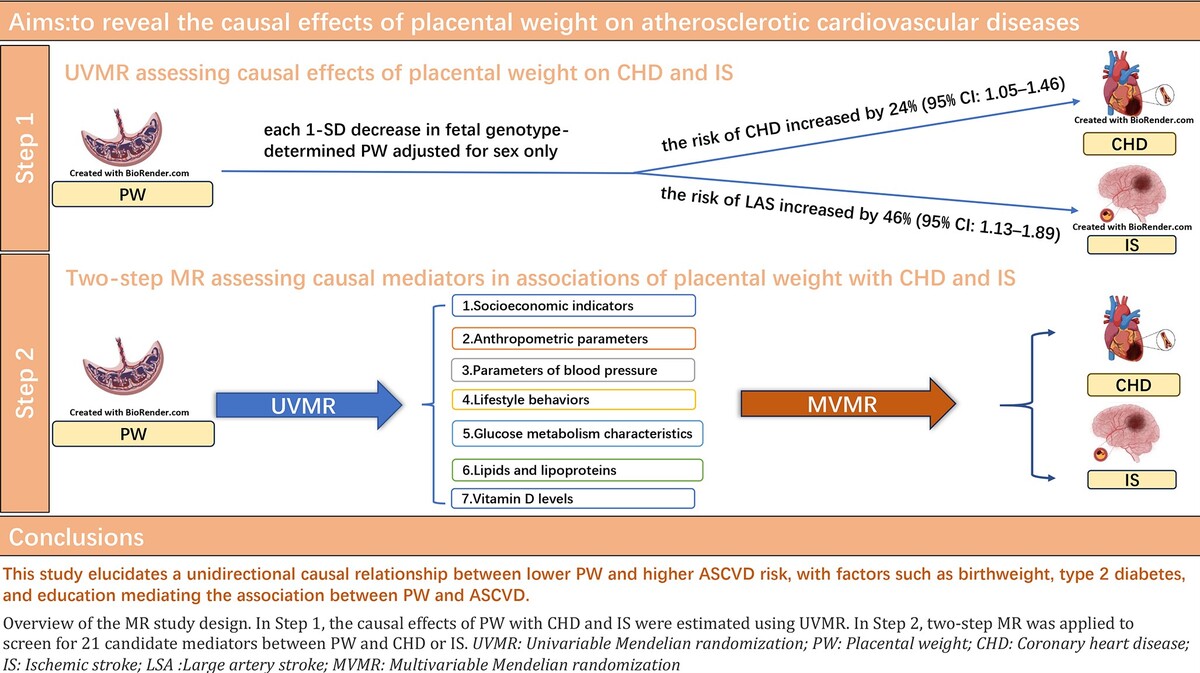
Using summary data from genome-wide association studies (GWAS) of PW, CHD, IS and 21 candidate mediators, we established a workflow to examine the role of these traits as plausible mediators of the effects of PW on CHD and IS risk. Specifically, this MR study consisted of two phases of analysis. In the first phase, we used bidirectional two-sample univariate MR (UVMR) to assess the causal effect of PW on ASCVD (Figure 1 A). In the second phase, we screened 21 candidate mediators that may mediate the association between PW and ASCVD and calculated the proportion of mediators for each eligible mediator using two-step MR (Figure 1 B).
Figure 1
A – Overview of the MR study design. This MR study consisted of two analysis phases. In Phase 1, the causal associations of PW with CHD and IS were estimated using bidirectional UVMR. B – In Phase 2, two-step MR was applied to screen for 21 candidate mediators that may mediate the association between PW and ASCVD and quantify the mediation proportions for qualified mediators
UVMR – univariable Mendelian randomization, MVMR – multivariable Mendelian randomization, PW – placental weight, EGG – early growth genetics, CHD – coronary heart disease, IS – ischemic stroke, ASCVD – atherosclerotic cardiovascular disease, IVs – instrumental variables.
We used multiple approaches to fulfill the three core assumptions of MR as follows [29]. First, the IVs are strongly associated with at least one of the exposures (i.e., PW) in UVMR analyses or at least one of the multiple exposures in multivariable MR (MVMR) analyses. Second, IV was independent of confounders of the relationship between exposure and outcome (i.e., CHD and IS). Third, IVs affect outcomes only through exposure, not through any direct or indirect pathways. This study used summary statistics from publicly available GWAS from large-scale studies of individuals of predominantly European ancestry. This MR study was reported according to the Guidelines for Strengthening the Reporting of Observational Studies in Epidemiology Using Mendelian Randomization (STROBE-MR) [34].
The dataset containing all the data used in this study is publicly accessible on the database website. The Institutional Review Board’s Ethics Committee duly authorized written informed consent from all participants. Therefore, no additional ethical approval or informed consent was needed.
Data sources for and selection of genetic instrumental variables
The sources of exposure, mediator factors, and outcome data used in this study are provided in the supplementary materials (Supplementary Table SII).
Placental weight
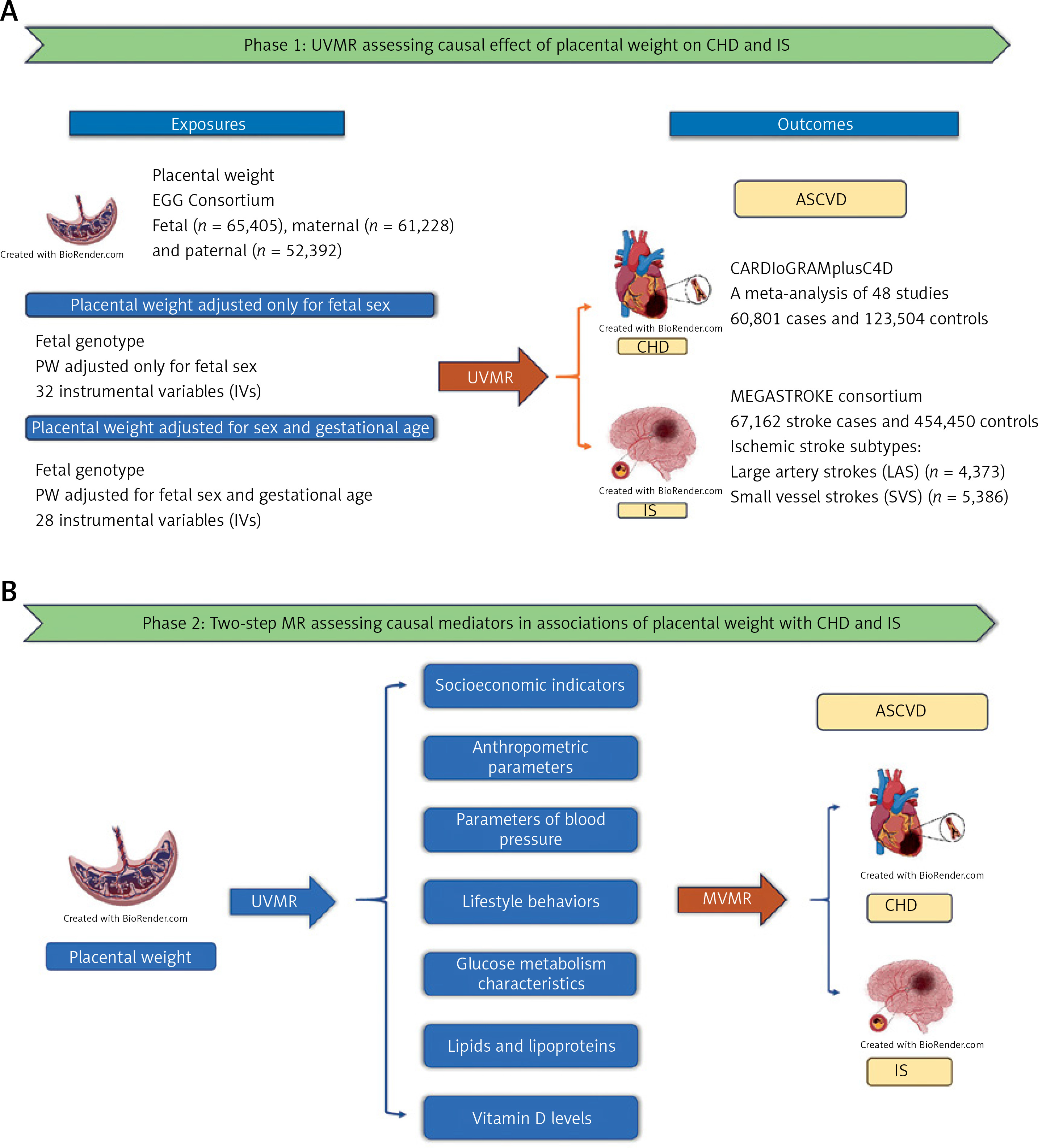
Summary statistics for PW are available from the Early Growth Genetics (EGG) Consortium’s latest GWAS for PW. This study conducted a GWAS meta-analysis of fetal genotype (n = 65,405), maternal genotype (n = 61,228) and paternal genotype (n = 52,392) data from 21, 16, and 6 European studies according to fetal sex and gestation duration, respectively [35]. PW information was collected from medical records, researcher measurements, records at delivery, and birth registries. In the EGG consortium, PW data were converted to z scores for analysis via the within-cohort, unadjusted PW mean and standard deviation to calculate z scores for each individual. Individuals reporting multiple pregnancies, congenital anomalies, gestational age < 37 weeks and > 43 weeks, PW < 200 and > 1500 g, and z scores ±5 SDs were excluded [35].
The study assessed the association between each genetic variant and PW using linear regression or linear mixed models, adjusted for fetal sex, and identified 45 autosomal single-nucleotide polymorphisms (SNPs) independently associated with PW (p < 5 × 10–8). Of these, 39 association loci were identified by fetal GWAS meta-analysis (p < 5 × 10–8), 4 loci by maternal analysis, and 2 loci by paternal analysis [35]. In addition, the study conducted a GWAS meta-analysis adjusted for fetal sex and gestational age. A total of 41 loci reached genome-wide significance (p < 5 × 10–8), of which 32 association loci were identified by fetal GWAS meta-analysis (p < 5 × 10–8), 4 loci by maternal analysis, and 2 loci by paternal analysis [35]. We performed more stringent linkage disequilibrium clumping with a threshold r2 < 0.001 cut-off using a 1,000-genome reference panel within a 10,000 kb window to select independent genetic variants. Finally, in analyses adjusted for fetal sex, we identified 32 IVs of fetal origin, 3 IVs of maternal origin, and 1 IV of paternal origin. There were 28 fetal-origin IVs, 3 maternal-origin IVs, and 2 paternal-origin IVs in the analyses adjusted for fetal sex and gestational age. Considering that the small number of validated IVs for paternal and maternal sources may lead to low study power in Mendelian randomization studies, in the present study we performed Mendelian randomization analyses using 32 IVs of fetal origin adjusted for fetal sex. As a supplement, we used 28 IVs of fetal origin adjusted against fetal sex and gestational age for repeated analyses (Supplementary Table SIII).
Candidate mediators
Based on a literature review of previous observational and MR studies, we focused on 21 candidate regulatory mediators that may be present in the association between PW and ASCVD (Supplementary Table SIV), all of which can be found in available GWAS genetic tools. The 21 candidate mediators were clustered into seven groups as follows: (1) socioeconomic indicators including education and household income; (2) anthropometric parameters including birthweight, body mass index (BMI), and waist-to-hip ratio (WHR); (3) blood pressure parameters including hypertension, systolic blood pressure (SBP) and diastolic blood pressure (DBP); (4) lifestyle behaviors including cigarettes smoked per day, alcoholic drinks per week and coffee consumption; (5) glucose metabolism parameter including type 2 diabetes, glycated hemoglobin (HbA1c); (6) lipids and lipoproteins including total cholesterol-high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), triglycerides, apolipoprotein B, omega-3 fatty acids, omega-6 fatty acids; (7) vitamin D levels (Supplementary Table SIV).
In UVMR analyses, genetic IVs for each candidate mediator were genome-wide significant (p < 5 × 10–8) and independent of each other (LD r2 < 0.001 within 10,000 kb). In MVMR analyses, genetic IV was a combination of SNPs that were genome-wide significant (p < 5 × 10–8) and independent of each other (LD r2 < 0.001 within 10,000 kb) in either the GWAS for birthweight or the GWAS for each candidate mediator variable. If there were no SNPs for exposures in the GWAS summary statistics for CHD or IS, we used SNPs with chain disequilibrium (r2 > 0.8) as proxies.
We screened for mediators of the causal associations of PW with CHD and IS according to the following criteria: [1] PW should be causally related to the mediator variables; [2] the mediator variables should have a direct causal effect on the outcome, independent of PW; and [3] the mediating effect of the mediator variables should be in the same direction as the total effect of PW on the outcome.
Outcomes
GWAS summary statistics for CHD were obtained from the UK Biobank and the Coronary Artery Disease Genome-wide Replication and Meta-analysis Consortium (CARDIoGRAM). The CARDIoGRAMplusC4D 1000 genome-based GWAS is a meta-analysis of 48 studies, including data from 60,801 cases of coronary artery disease and 123,504 controls of white European ancestry. Coronary heart disease was diagnosed according to the International Classification of Diseases (ICD-10: I20, I21, I22) [36].
Unlike CHD, the etiology of IS is diverse, and in addition to atherosclerotic embolization, other etiologies include cardiac embolization, small-artery atherosclerosis, and other mechanisms. The ischemic stroke subtypes associated with atherosclerosis include two main types: large artery stroke (LAS) and small vessel stroke (SVS). The GWAS summary statistics for IS and its subtypes used in this study were obtained from the MEGASTROKE Consortium, with a total of 34,217 cases of any ischemic stroke of European ancestry and 406,111 controls of European ancestry, including 4,373 cases of LAS and 5,386 cases of SVS [37]. Ischemic stroke and its two subtypes (large-artery stroke and small-vessel stroke) were classified according to the Org 10172 trial in the Treatment of Acute Stroke (TOAST) system [38].
Statistical analysis
UVMR and MVMR analyses
We used the inverse variance weighted (IVW) method as the primary analysis for UVMR and the multivariate inverse variance weighted (MV-IVW) method as the primary analysis for MVMR. The IVW method uses a random-effects meta-analysis to combine the Wald ratio estimates for each SNP in the IV group into a single causal estimate [39]. MR causal estimates were provided as odds ratios (ORs) (95% CIs) for binary outcomes and as β coefficients (95% CIs) for continuous outcomes.
Total effects of PW on CHD and IS
We performed bidirectional UVMR to assess the overall causal effects of placental weight on CHD and IS, an approach that excludes confounding effects due to reverse causal associations. In addition, we performed leave-one-out analyses to assess the effects of individual variants on these associations. This approach examined whether specific genetic variants dominated estimates of causal effects by excluding one valid SNP at a time in IVW analyses.
Mediation MR analysis
We applied two-step MR analysis to assess whether mediators could mediate the associations between PW and ASCVD, respectively [40]. The first step was to estimate the causal effect of genetically determined PW on each potential mediator (β1) using UVMR. The second step was to estimate the causal effect of each potential mediator on CHD or IS (β2) using MVMR with adjustment for PW. Where there was evidence that PW affected the mediator variable, which in turn affected the outcome, we used a ‘product of coefficients’ approach to assess PW’s mediating effect on ASCVD via each mediator variable (β1 × β2). The mediation proportion for each mediating variable was calculated by dividing the mediated effect by the total effect. The standard errors of the mediation effects were calculated via the delta method. We set the lower limit of the negative mediation proportion to 0% because this is the minimum threshold for determining the mediation proportion.
MR sensitivity analysis
For UVMR analysis, we used the inverse variance weighting (IVW) method as the primary analysis and adopted the MR-Egger, weighted median, simple mode, weighted mode, and MR polytomous residual sums and outliers (PRESSO) methods as sensitivity analyses of UVMR to assess the robustness of IVW estimates under different assumptions. The MR-Egger method allows for the free estimation of the intercept as an indicator of pleiotropy to identify and adjust for potential directional pleiotropy bias but with limited precision [41]. The weighted median method selects the median MR estimate as the causal estimate and provides a consistent causal estimate if more than 50% of the weights in the analysis are derived from valid IVs [42]. Simple and weighted mode methods cluster SNPs based on the similarity of causal effects and estimate causal effects on the basis of the maximum clustering of SNPs [43]. The presence of pleiotropy was also assessed by applying the MR-PRESSO method, which detects and corrects for any peripheral SNPs reflecting possible pleiotropy bias in MR causal estimates and assesses whether excluding peripheral SNPs affects causal estimates [44]. In addition, we used the cML-MA method for additional sensitivity analyses. cML-MA is an MR method based on constrained maximum likelihood and model averaging that has been used to control for both correlated and uncorrelated multicollinearity effects and is considered much more powerful than MR Egger [45].
For the MVMR analysis, we implemented the MVMR Egger method to verify the robustness of the MV-IVW results. The multivariate MR-Egger method has advantages over univariate methods in terms of the plausibility of the assumption required for consistent causal estimation and the ability to detect causal effects when the assumption is met [46]. We used the F-statistic to assess the validity of the IV and applied Cochran’s Q-statistic to assess heterogeneity. In addition, MR Egger’s intercept was used to assess the pleiotropy of the IVW estimates.
Given that multiple candidate mediators were tested in the analyses, we used the Benjamini-Hochberg method for false discovery rate (FDR) correction to account for multiple testing. IVW estimates with p < 0.05 and FDR q- values < 0.05 were regarded as evidence of causality, whereas IVW estimates with p < 0.05 but FDR q- values ≥ 0.05 were regarded as suggestive causal evidence.
All the statistical analyses and data visualizations were performed using the R packages “TwoSample MR”, “MRPRESSO”, and “MRcML”. These packages are readily available in R software (version 4.3.2).
Results
Causal associations of PW with CHD and IS
In the UVMR analysis of PW with CHD (PW adjusted for sex only), a lower fetal genotype-determined PW was positively associated with a higher risk of CHD (IVW - OR = 1.24; 95% CI: 1.05–1.46; p = 1.10 × 10–2). The same conclusion was reached in the repeat UVMR analysis of PW with CHD (PW adjusted for gestational duration and sex) (IVW – OR = 1.20; 95% CI: 1.00–1.43; p = 4.90 × 10–2) (Figure 2 A; Supplementary Table SV).
Figure 2
A – UVMR estimates (IVW) for the causal associations of PW with CHD and IS. The causal associations of PW with CHD. ORs (95% CIs) represent the risk for CHD or IS associated with each 1-SD lower PW. B – The causal associations of PW with IS. ORs (95% CIs) represent the risk for CHD or IS associated with each 1-SD lower PW
UVMR – univariable Mendelian randomization, IVW – inverse variance weighted, PW – placental weight, CHD – coronary heart disease, IS – ischemic stroke, OR – odds ratio, SD – standard deviation.
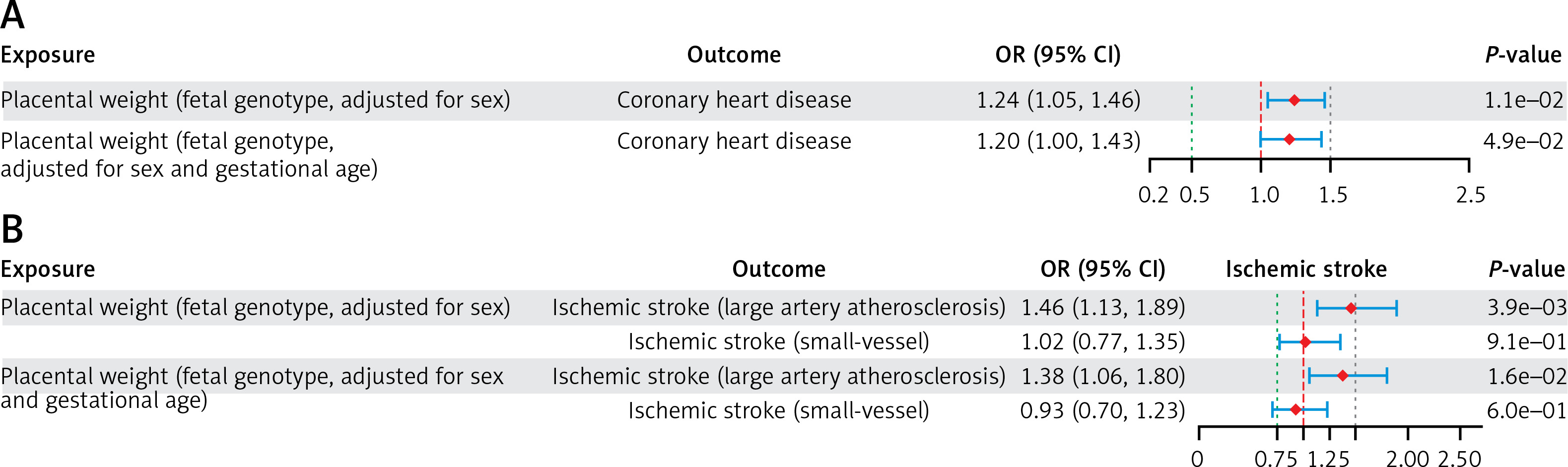
In the UVMR analysis of PW with IS (PW adjusted for sex only), a lower fetal genotype-determined PW was positively correlated with a higher risk of LAS (IVW – OR = 1.46; 95% CI: 1.13–1.89; p = 3.9 × 10−3). The same conclusion was reached in the repeated UVMR analysis of PW with IS (PW adjusted for gestational duration and sex) (IVW - OR: 1.38; 95% CI: 1.06–1.80; p = 1.6 × 10–2) (Figure 2 B; Supplementary Table SVII). In contrast, no correlation was found between PW and SVS risk in the UVMR analysis of PW with IS (Figure 2 B; Supplementary Table SVII).
In the reverse MR analysis, no causal associations were found between CHD and PW or between IS and PW (Supplementary Tables SVI and SVIII).
The above IVW estimates have been confirmed by at least one sensitivity analysis. MR-Egger intercept analysis did not reveal evidence of horizontal pleiotropy (all p intercepts ≥ 0.05; Supplementary Tables SIX and SX). Cochran’s Q test indicated potential heterogeneity, but IV validity revealed that instrument strength was sufficient (all SNP F-statistics ≥ 10). Leave-one-out analysis revealed that no individual SNP significantly altered causal associations (Supplementary Figures S1 and S2).
Two-step MR assessing causal mediators in associations of PW with CHD and LAS
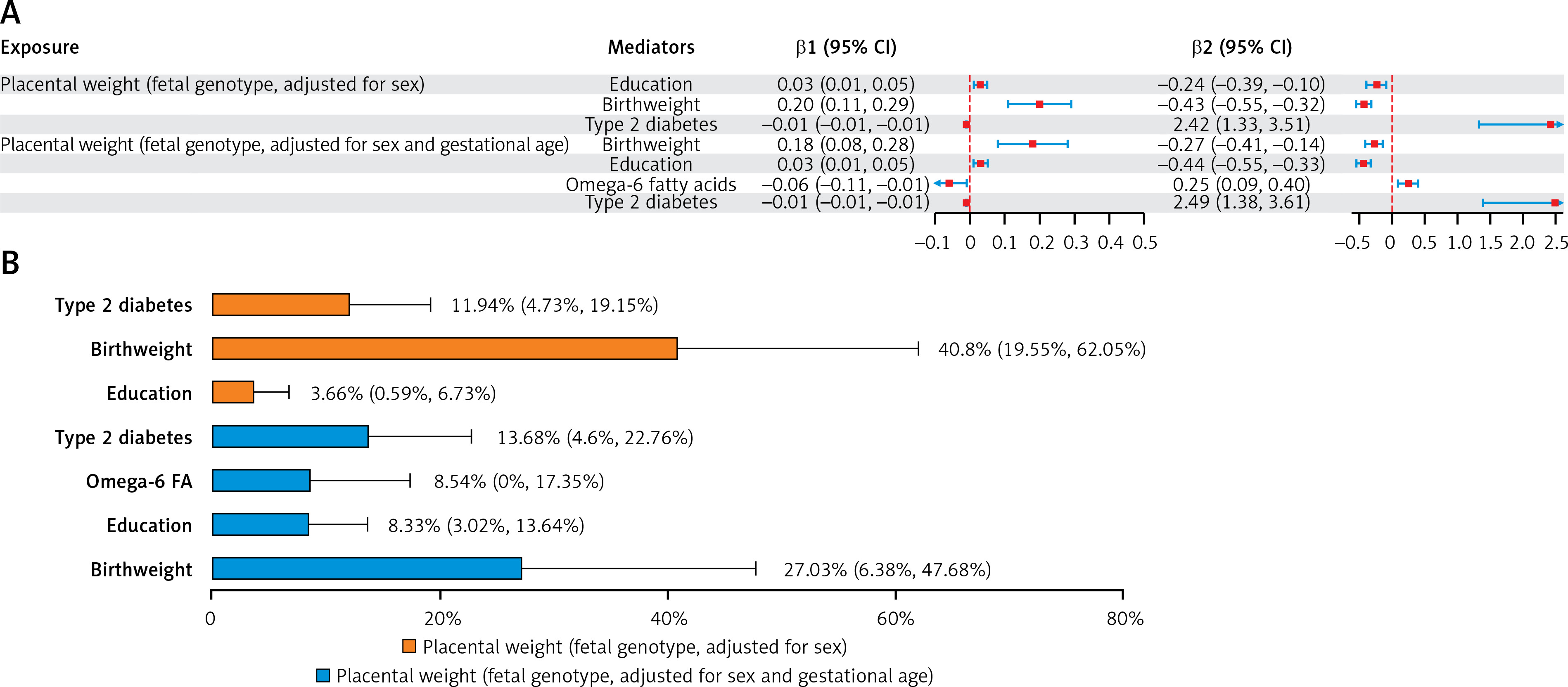
In the mediation analysis of PW with CHD (PW adjusted for sex only), out of 21 candidate mediators, birthweight, education and type 2 diabetes met all screening criteria, qualifying as mediators between fetal genotype-determined PW and CHD. In the repeated mediation analysis of PW with CHD (PW adjusted for gestational age and sex), birthweight, education, omega-6 fatty acid levels, and type 2 diabetes met all screening criteria, qualifying as mediators between fetal genotype-determined PW and CHD (Figure 3 A).
Figure 3
A – Selection process for mediators in the causal associations of PW with CHD and IS. The qualified mediators in the mediation analysis of PW with CHD. [1] PW should be causally related to the mediator variables; [2] the mediator variables should have a direct causal effect on the outcome, independent of PW; and [3] the mediating effect of the mediator variables should be in the same direction as the total effect of PW on the outcome. B – The qualified mediators in the mediation analysis of PW with LAS. Three criteria were applied to screen for the mediators in the causal associations of PW with CHD and IS
BMI – body mass index, WHR – waist-to-hip ratio, SBP – systolic blood pressure, DBP – diastolic blood pressure, HbA1c –glycated hemoglobin, HDL-C – high-density lipoprotein cholesterol, PW – placental weight, CHD – coronary heart disease, IS – ischemic stroke.
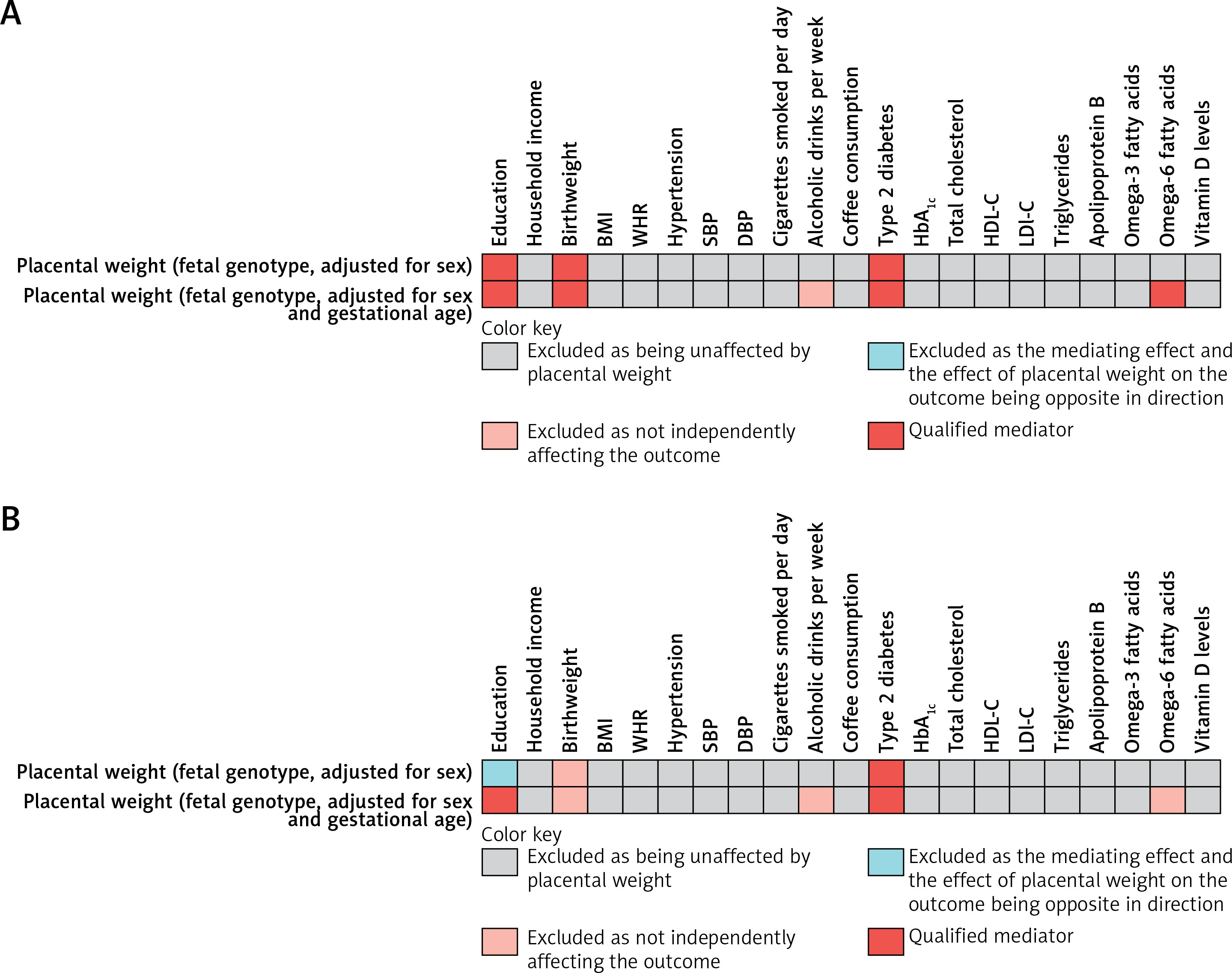
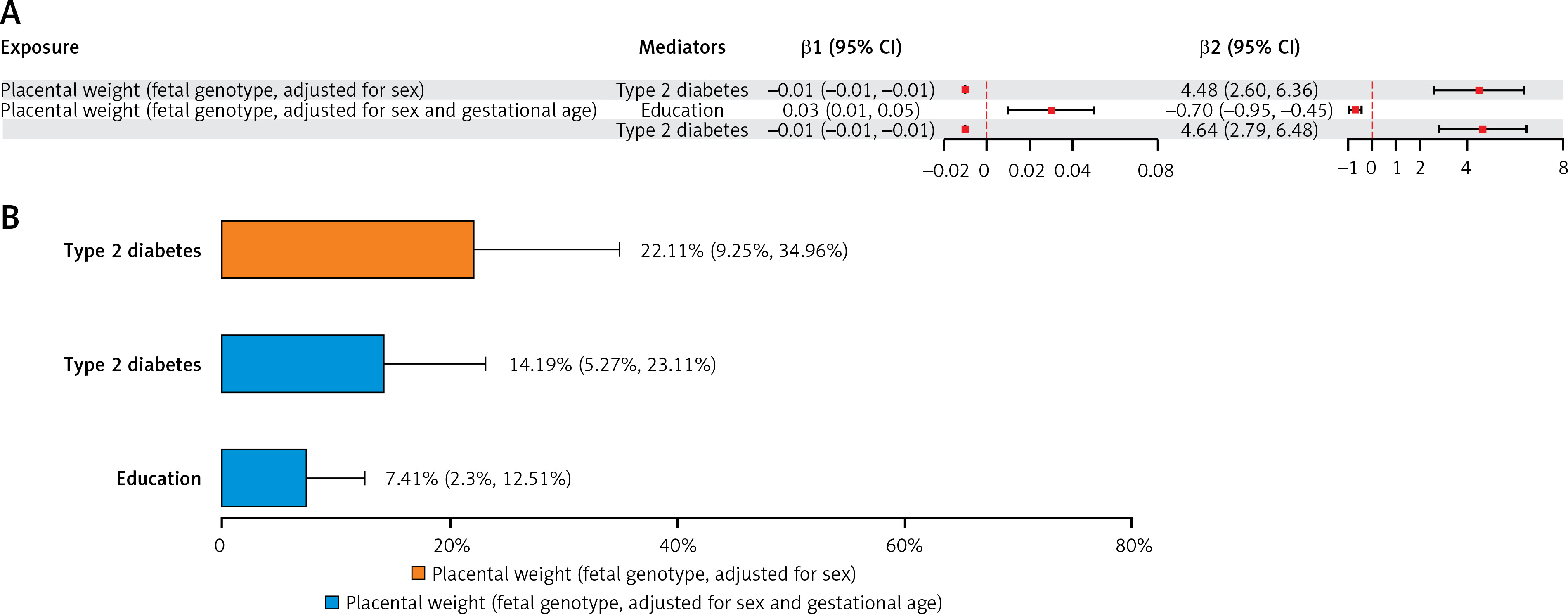
In the mediation analysis of PW with LAS (PW adjusted for sex only), among 21 candidate mediators, type 2 diabetes met all screening criteria. It was eligible as a mediator between fetal genotype-determined PW and LAS. In the repeated mediation analysis of PW with LAS (PW adjusted for gestational age and sex), education and type 2 diabetes met all screening criteria and were eligible as mediators between fetal genotype-determined PW and LAS (Figure 3 B).
Causal associations of PW with candidate mediators
In UVMR analyses of PW with mediators (PW adjusted for sex only), after false discovery rate (FDR) correction, a higher fetal genotype-determined PW was found to be causally associated with higher birthweight and educational attainment, as well as lower risk of type 2 diabetes (Supplementary Table SXI).
In the repeated UVMR analyses of PW with mediators (PW adjusted for gestational age and sex), higher fetal genotype-determined PW was found to be causally associated with higher alcohol intake, higher birthweight, and education attainment, as well as lower risk of type 2 diabetes, and lower levels of omega-6 fatty acid levels, after correction for FDR (Supplementary Table SXII).
Causal associations of mediators with CHD and LAS
In MVMR analyses of PW with CHD (PW adjusted for sex only), with adjustment for fetal genotype-determined PW, each 1-unit increase in genetically determined education and birthweight was associated with a 35% and 22% lower risk of CHD, respectively. In contrast, with each 1-unit increase in genetically determined type 2 diabetes, the CHD risk increased by 1025% (Supplementary Table SXIII). In the repeated analysis of PW with CHD (PW adjusted for gestational age and sex), with adjustment for fetal genotype-determined PW, each 1-unit increase in genetically determined education and birthweight was associated with a 36% and 24% lower risk of CHD, respectively. In contrast, each 1-unit increase in genetically determined omega-6 fatty acids and type 2 diabetes was associated with a 28% and 1108% higher risk of CHD, respectively (Supplementary Table SXIV).
In MVMR analysis of PW with LAS (PW adjusted for sex), with adjustment for fetal genotype-determined PW, for each 1-unit increase in genetically determined education, the risk of LAS decreased by 50%, and a causal relationship between genetically determined type 2 diabetes and increased risk of LAS was also found (Supplementary Table SXIV). In the repeated analysis of PW with LAS, with adjustment for fetal genotype-determined PW, for each 1-unit increase in genetically determined education, the risk of LAS decreased by 50%, and genetically determined type 2 diabetes was similarly shown to be causally associated with a higher risk of LAS (Supplementary Table SXIV).
Mediators in the associations of PW with CHD and LAS
In the mediation analysis of PW with CHD (PW adjusted for sex), birthweight, type 2 diabetes, and education mediated 40.80%, 11.94%, and 3.66%, respectively, of the total effect of fetal genotype-determined PW on CHD. In the repeated analysis of PW with CHD (PW adjusted for gestational age and sex), birthweight, type 2 diabetes, omega-6 fatty acids, and education mediated 27.03%, 13.68%, 8.54%, and 8.33%, respectively, of the total effect of fetal genotype-determined PW on CHD (Figure 4; Supplementary Table SXV).
Figure 4
A – The mediating role of each mediator in the causal associations of placental weight with coronary heart disease. The mediating effect (β1 × β2) of each qualified mediator in the causal associations of PW with CHD. B – Proportion of mediation mediated by each qualified mediator for coronary heart disease. Two-step MR was used to evaluate the mediating role of each mediator in the causal associations of placental weight with coronary heart disease
β1 – Effect of placental weight on the mediator. β2 – Effect of the mediator on coronary heart disease. PW – placental weight. CHD – coronary heart disease.
In the mediation MR analysis of PW with LAS, type 2 diabetes mediated 22.11% of the total effect of fetal genotype-determined PW on LAS. In the repeated analysis of PW with LAS, type 2 diabetes and education each mediated 14.19% and 7.41% of the total effect of fetal genotype-determined PW on LAS (Figure 5; Supplementary Table SXVI).
Figure 5
A – The mediating role of each mediator in the causal associations of placental weight with large artery stroke. The mediating effect (β1 × β2) of each qualified mediator in the causal associations of PW with large artery stroke. B – Proportion of mediation mediated by each qualified mediator for large artery stroke. Two-step MR was used to evaluate the mediating role of each mediator in the causal associations of placental weight with large artery stroke
β1 – Effect of placental weight on the mediator, β2 – Effect of the mediator on large artery stroke, PW – placental weight, IS – ischemic stroke.
Discussion
This MR study first evaluated the independent causal effect of low PW on ASCVD risk, identified potential mediators, and quantified their mediating role in this relationship. Specifically, in the UVMR analysis of PW with CHD, for every 1-SD decrease in fetal genotype-determined PW, the risk of CHD increased by 24%. Similar results were obtained in the repeated analysis (for every 1-SD decrease in PW, CHD risk increased by 20%). In the UVMR analysis of PW and IS, for every 1-SD decrease in fetal genotype-determined PW, the risk of large artery stroke (LAS) increased by 46%. Similar results were obtained in the repeated analysis of PW and IS (for every 1-SD decrease in fetal genotype-determined PW, the LAS risk increased by 38%).
In the UVMR analysis of PW and IS and the repeated analysis, no correlation was found between PW and the risk of small vessel stroke (SVS). We believe that this is because the pathological features of small vessel stroke differ from those of LAS [47]. Atherosclerosis is not a major pathological feature of small-vessel stroke. Notably, the risk factors for small-vessel stroke as a whole also differ from the typical proatherogenic risk factors for large-artery stroke [48].
The mediation MR analysis and repeated analyses revealed that the causal association between lower placental weight and CHD risk was substantially mediated by birth weight, type 2 diabetes, and education. On the other hand, type 2 diabetes played a primary mediating role in the association between lower placental weight and LAS.
The intrauterine development stage is critical for lifelong health because the foundation of body planning and major organ systems is laid during this period. Anatomically, the placenta connects the fetus to the mother, transfers nutrients from the mother to the fetus, and creates a stable environment for fetal development, ensuring isolation from maternal and environmental stressors [9]. However, if normal placental function is compromised or exceeds the organ’s adaptive capacity, the fetal environment may be disrupted, significantly impacting the lifelong health of the offspring [9]. Like low birthweight, low placental weight represents intrauterine malnutrition [9]. Low placental weight indicates poor placental growth during pregnancy, which can restrict the flow of nutrients and contribute to fetal malnutrition [9]. Previous studies have shown that low birth weight is associated with an increased risk of cardiometabolic disease later in life [9, 28, 49–51]. However, research on the causal relationship between low placental weight and the subsequent development of cardiovascular disease in later life is still lacking. Our MR study elucidated the causal relationship between low placental weight and atherosclerotic cardiovascular disease. Our findings can be interpreted by the thrifty phenotype hypothesis. The adaptation of individuals to a nutrient-deficient intrauterine environment may result in modifications to insulin signaling pathways, epigenetic alterations, and variations in the proliferation and differentiation of cells within essential organs. These changes can subsequently lead to disturbances in the metabolism of glucose, lipids and other metabolites, ultimately predisposing individuals to cardiometabolic diseases as adults [6, 52]. From a clinical perspective, our study indicated that individuals with low placental weight have a higher risk of developing ASCVD, encouraging further consideration of PW in risk stratification for ASCVD.
This MR study offered novel and reliable evidence on the causal link between low PW and late-life ASCVD risk, highlighting birthweight, type 2 diabetes, and education level as potential mediators. The strengths of this study included the rigorously designed MR analysis framework to investigate the causal relationship and potential mediating factors between low PW and ASCVD, and we used a substantial amount of GWAS summary statistics data. However, this study also has several limitations. First, this MR study was almost entirely restricted to individuals of European descent to ensure consistency in genetic background. The use of GWAS summary data mainly composed of individuals of European descent may introduce biased estimates and limit generalizability to different racial populations. Second, based on a two-sample MR design, we assumed that the relationship between the PW and outcomes in UVMR and MVMR analyses is linear. Future research is necessary to investigate potential non-linear causal relationships using individual-level data. Third, potential exposure-mediator interactions cannot be modeled in the current two-sample MR setting. Nonetheless, the MR approach could largely alleviate the potential bias caused by the exposure-mediator interaction [50, 53]. Fourth, to facilitate clinical practice, we focused on theoretically common and important candidate mediators associated with ASCVD as candidate mediators. However, this study cannot fully capture all the mediating factors, especially non-genetic factors. Finally, PW measured postpartum only roughly represents placental growth status and cannot entirely directly represent placental injury or placental dysfunction; future research needs to introduce more relevant indicators.
In conclusion, our study revealed a causal relationship between genetically predicted lower placental weight and a greater risk of ASCVD. Mediators such as birth weight, type 2 diabetes, and education partially mediated the association between low placental weight and ASCVD. This finding underscores the significance of low PW as a predictor and a preventive factor for late-life ASCVD risk and offers substantial insights into the developmental origins of health and disease (DOHaD) hypothesis.