Introduction
Cholesterol is a lipid molecule that plays a crucial role in the numerous physiological processes of the body. While it serves many significant functions, excessive cholesterol can also lead to serious medical conditions such as coronary heart disease. This review aims to provide an overview on cholesterol and its sources, metabolism, functions, and potential health consequences. Through an examination of current literature and research, this review synthesizes information to provide a comprehensive description of cholesterol. According to cholesterol metabolism, cholesterol derives from two sources: endogenous generation (produced within the body) and external intake (obtained through diet). The liver is the primary location of endogenous cholesterol synthesis, and numerous metabolic processes, including the mevalonate system, contribute to its synthesis. Exogenous cholesterol, on the other hand, is predominantly derived from animal foods such as meat, eggs, and dairy products [1].
Cholesterol metabolism is the pathway via which cholesterol is synthesized, absorbed, transported, and eliminated from the body. Cholesterol is produced in the liver through a series of complex enzyme reactions via the mevalonate pathway under the control of various factors including genetics and hormones [2]. Most mammalian cells can synthesize cholesterol from acetyl-CoA. Three acetyl-CoA molecules are condensed to form 3-hydroxy-3-methyl-glutaryl-CoA (HMGCoA), which is then transformed to mevalonate by HMG-CoA reductase. Intestinal cells absorb dietary cholesterol and transport it either as free cholesterol or as part of lipoproteins to the bloodstream. Cholesterol elimination occurs chiefly through sterol and bile acid excretion. Mammalian cells absorb cholesterol from the bloodstream through the low-density lipoprotein (LDL) receptor (LDLR), in addition to de novo production. LDLs are created from VLDLs produced by the liver, which gradually release their triglyceride content. LDLR-mediated endocytosis of cholesterol-rich LDL transports the complex to the lysosome for degradation and cholesterol release [3]. Endogenous cholesterol synthesis in normal healthy adults occurs at a rate of about 1 g/day, with dietary consumption contributing an extra 0.3 g/day. Despite these inputs, the body maintains relatively consistent plasma cholesterol levels, which range between 150 and 200 mg/dl, according to finely regulated homeostatic systems [4]. The rate of de novo cholesterol synthesis, which occurs predominantly in the liver, is an important factor in maintaining this balance and is influenced in part by dietary cholesterol consumption, ensuring that total cholesterol (TC) levels remain within a physiologically acceptable range [5].
Cholesterol serves several critical body functions. It is a structural unit of cell membrane structure, providing strength to them and maintaining them in a fluid state [6–8]. Moreover, cholesterol is a precursor for steroid hormone biosynthesis, bile acid biosynthesis, and vitamin D biosynthesis [9]. These activities indicate the irreplaceable role cholesterol plays in maintaining normal body function. Cholesterol homeostasis is strictly controlled to avoid the cytotoxic effects of excess free cholesterol. One immediate physiological response to cholesterol overload is the enzymatic conversion of cholesterol to cholesteryl esters by acyl-CoA:cholesterol acyltransferase (ACAT). These inert esters are temporarily retained in intracellular lipid droplets to provide short-term protection against cholesterol buildup. However, due to the limited storage capacity of lipid droplets, this method is insufficient in the presence of chronic cholesterol excess. To maintain long-term homeostasis, cells must swiftly inhibit endogenous cholesterol manufacture and absorption while boosting the outflow of excess cholesterol for disposal. This coordinated response ensures that excess cholesterol is eventually eliminated from the body, primarily through the fecal pathway, protecting cells from lipid-induced toxicity [10].
Alterations in lipid organization within cellular membranes can significantly influence key processes such as signal transduction and membrane trafficking. These disruptions may arise from genetic mutations, environmental influences (for example, diet), or a combination of both, and are increasingly recognized as contributing factors to human disease. While cholesterol is necessary for cellular structure and function, abnormal levels can have negative health repercussions [11]. Dysregulated cholesterol is widely recognized as a major risk factor for cardiovascular disorders; nevertheless, new evidence links cholesterol to the pathogenesis of a variety of ocular diseases, including age-related macular degeneration (AMD), cataracts, glaucoma, and retinal vascular disorders [12]. A study of Chinese adults by Xia et al. (2023) [13] found that 42.1% had dyslipidemia, with low HDL-C being the most common subtype, followed by high triglycerides (TG) (15.4%), high TC (8.3%), and high LDL-C (7.1%). Understanding the mechanisms underlying cholesterol metabolism and eye illness is critical for creating effective preventive and treatment measures. This review explores the relationship between cholesterol homeostasis and ocular health, with particular emphasis on the role of cholesterol in the development and progression of common vision-threatening disorders. Unless otherwise specified, this review refers to cholesterol-related pathology primarily reflecting LDL-C-driven lipid dysregulation, while also considering the modifying and protective roles of HDL-C and the contribution of hypertriglyceridemia. Accordingly, the scope of this review focuses on lipid metabolism and lipoprotein imbalance, rather than TC alone.
Literature search strategy
A comprehensive and structured literature search was conducted to identify relevant studies examining the role of cholesterol and lipid metabolism in ocular diseases. Electronic databases including Scopus, Web of Science, and Google Scholar were systematically searched for articles published between January 2000 and June 2025. This time frame was selected to capture foundational mechanistic studies as well as recent advances in molecular pathways, clinical associations, and emerging therapies.
Search terms were developed using a combination of Medical Subject Headings (MeSH) and free-text keywords related to cholesterol biology and eye diseases. Core search terms included cholesterol, lipid metabolism, lipoproteins, LDL, HDL, dyslipidemia, mevalonate pathway, SREBP, ABCA1, oxidative stress, and inflammation, combined with ocular-related terms such as AMD, diabetic retinopathy, retinal vascular disease, cataract, and glaucoma. Boolean operators (AND and OR) were applied to refine searches.
Original research articles, clinical studies, systematic reviews, and meta-analyses published in English within the specified period were included. Reference lists of eligible articles were manually screened to identify additional relevant studies, with emphasis placed on recent publications (2018–2025) to ensure contemporary clinical and translational relevance.
Cholesterol and its role in the human body
General functions of cholesterol
Lipid composition affects membrane characteristics, and cholesterol plays an important role in this process by regulating membrane fluidity and permeability while also inducing the development of coexisting phases and domains in the membrane. Cholesterol molecules are dispersed between the phospholipid bilayer, regulating membrane fluidity and permeability [14]. Furthermore, cholesterol promotes the synthesis of complex membrane microdomains known as lipid rafts, which play important roles in cell signaling and membrane trafficking [11].
Cholesterol not only serves as a molecule of regulation itself but also forms the backbone of all steroid hormones and vitamin D analogs. Cholesterol is also required for the synthesis of steroid hormones such as cortisol, aldosterone, and sex hormones such as progesterone, estrogen, and testosterone [15]. Cholesterol serves as an obligatory precursor for steroid hormone production in steroidogenic tissues, such as the adrenal glands, gonads, placenta, and brain. Regardless of the tissue of origin or the steroid hormone to be produced, steroidogenesis is initiated by the cleavage of the cholesterol side chain to produce pregnenolone. The protein catalyzing this reaction, P-450 side chain cleavage (P450scc/CYP11A1) enzyme, is located in the inner mitochondrial membrane [16]. These hormones are involved in many of the body’s physiological processes, including response to stress, electrolyte balance, and sexual growth and reproduction [15, 16].
Neurosteroids are steroids that are produced in the brain itself. They are multifunctional lipids with effects mediated by ion-gated neurotransmitter receptors rather than typical steroid hormone nuclear receptors. In addition to modulating receptor activation, particular neurosteroids are known to regulate myelination, neuroprotection, and axon and dendritic development. Furthermore, cholesterol acts as a precursor to the production of bile acids, which aid in fat digestion and absorption [17].
Regulation of cholesterol levels
Dietary cholesterol is absorbed by intestinal enterocytes, whereas cholesterol synthesized in the liver can link to lipoproteins to create protein-lipid complexes, which are subsequently released into the bloodstream and transported to cells. In humans, enterocytes excrete roughly 25% of excess cholesterol straight into feces, with the remainder returning to the liver via reverse cholesterol transport (RCT) and excreted as bile. Only a small amount of cholesterol is reabsorbed and returned to the free cholesterol (FC) pool [18]. The mammalian body maintains cholesterol balance using a highly regulated system that ensures homeostasis.
The three-sterol regulatory element-binding proteins, SREBP1a, SREBP1c, and SREBP2, play critical roles in a variety of physiological and pathological processes, including fatty acid synthesis, endoplasmic reticulum (ER) stress, apoptosis, and autophagy, indicating their central role in lipid metabolism and synthesis [19]. SREBP cleavage-activating protein (SCAP), an ER membrane protein, is essential for controlling SREBP activity because it transports SREBPs from the ER to the Golgi apparatus, where proteases cleave SREBP and release its N-terminal domain. Once liberated, this N-terminal domain enters the nucleus and triggers the transcription of important lipogenic genes such as HMG-CoA reductase, HMG-CoA synthase, fatty acid synthase, and glycerol-3-phosphateacyltransferase [20].
SREBP-2 is particularly important among SREBPs because it acts as a liver-specific transcription factor, strictly regulating cholesterol homeostasis by modulating genes involved in cholesterol production and uptake [21]. In contrast, SREBP1c is the major transcription factor responsible for controlling hepatic de novo lipogenesis, particularly in response to insulin signaling. SREBP1c is originally generated as an inactive precursor anchored in the ER membrane; with insulin stimulation, it is proteolytically activated via the ER-to-Golgi trafficking pathway, which is also dependent on SCAP [22]. SCAP acts as a chaperone protein, promoting trafficking and permitting the proteolytic cleavage of SREBP, allowing its active domain to enter the nucleus and regulate lipogenic gene expression. When SCAP is suppressed, the activation process is disturbed, leading to decreased expression of enzymes involved in triglyceride and fatty acid synthesis, and hence reduced de novo lipogenesis [20, 22]. When intracellular cholesterol levels are low, SREBP-2 activates genes that control cholesterol synthesis, absorption, and conversion to bile acids. As cellular cholesterol levels rise, SREBP-2 activity decreases, limiting cholesterol formation and boosting its excretion from the body [23].
HMG-CoA reductase is regulated not just at the transcriptional level but also by post-translational mechanisms. When cholesterol levels in the ER rise, HMG-CoA reductase binds directly to cholesterol and the ER membrane-anchored ubiquitin ligase gp78, causing its destruction. This binding causes polyubiquitination of HMG-CoA reductase, which is then degraded via the 26S proteasome pathway. In contrast, when ER cholesterol levels are low, HMG-CoA reductase turnover is dramatically reduced due to decreased polyubiquitination and limited proteasome destruction [24]. Statins, the most often prescribed type of cholesterol-lowering medications, work by indirectly activating the SREBP regulatory system. They operate by competitively inhibiting HMG-CoA reductase, reducing ER cholesterol levels. This decrease in ER cholesterol induces SCAP-dependent activation of SREBPs, which increases the transcription of target genes involved in cholesterol absorption and synthesis. As a result, LDL receptor (LDLR) expression on the cell surface rises, resulting in better clearance of LDL particles from the circulation [25].
Impact of cholesterol on health
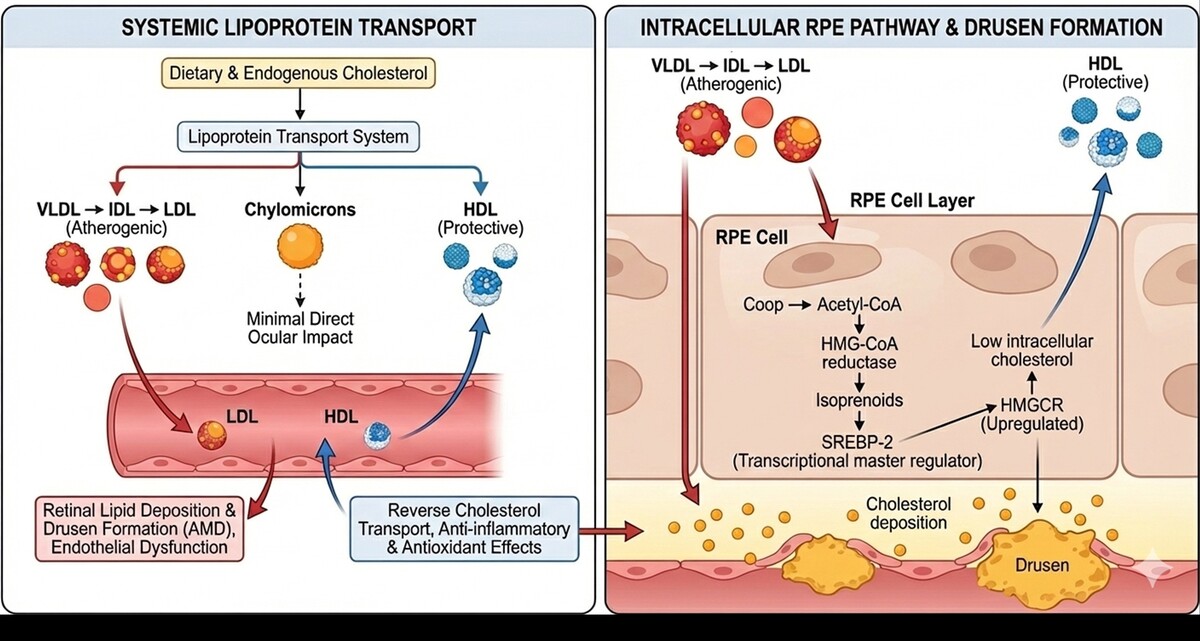
Lipoproteins, made up of unesterified cholesterol, TG, phospholipids, and proteins, circulate in the blood. There are five forms of lipoproteins: chylomicrons, very low-density lipoproteins (VLDL), intermediate-density lipoproteins (IDL), LDL, and high-density lipoproteins (HDL) [26]. Each has a role in delivering cholesterol and TG to various tissues. Cholesterol is required for optimal cellular function. However, abnormal cholesterol levels are closely associated with cardiovascular disease. LDL, also known as “bad cholesterol”, is a significant contributor to atherosclerosis, coronary artery disease, and stroke. In contrast, HDL or “good cholesterol” is linked to a lower risk of cardiovascular disease. A higher HDL/LDL ratio is thought to be a more reliable indicator of cardiovascular health than TC alone [27] (Figure 1).
Figure 1
Different types of cholesterol and their health implications. The figure delineates the pathways of dietary and endogenous cholesterol through the systemic lipoprotein transport system, highlighting the specific roles and pathological associations of various lipoproteins within the body
Excessive cholesterol, particularly free intracellular cholesterol, may be harmful. It builds up in cell organelles such as the plasma membrane, mitochondria, and ER. High cholesterol levels in macrophages enhance Fas ligand expression on the cell surface, causing mitochondrial death via the pro-apoptotic protein Bax. This promotes plaque instability in atherosclerosis. The cholesterol-induced cell death pathway involves either plasma membrane or ER stress, with the latter causing ER calcium depletion and activation of the unfolded protein response, which promotes cell death [28]. Free cholesterol can also crystallize inside cells, increasing toxicity and causing macrophage death. This mechanism increases the destabilization of atherosclerotic plaques, which can contribute to acute cardiovascular events [29]. Thus, maintaining cholesterol homeostasis is critical for preventing cardiovascular disease.
Cholesterol is an important structural component of cellular membranes, and recent diabetes research has shown that cholesterol levels in adipocytes, skeletal muscle fibers, and pancreatic β cells have a major impact on insulin sensitivity and secretion [30]. As a result, disturbances in cellular cholesterol homeostasis are increasingly thought to have a role in the development of type 2 diabetes (T2D). Recent clinical trials have found that patients with genetic variants that alter cholesterol metabolism are more likely to develop T2D. Furthermore, a series of recent studies in both animal models and human patients have shown that insulin-stimulated glucose uptake is inversely associated with plasma membrane cholesterol level, implying that increased membrane cholesterol reduces insulin action [31].
Cholesterol is essential for hormone synthesis, energy production, and cellular membrane integrity. However, excessive buildup in the body, particularly in organs and the bloodstream, is linked to an elevated risk of a variety of disorders, among them cancer [32]. Research has demonstrated that high cholesterol levels lead to cancer cell malignancy and progression [33, 34]. Mevalonate, a precursor in cholesterol production, increases breast cancer cell proliferation in both in vitro and in vivo studies [35, 36]. Additionally, estrogen receptor-positive breast cancer tissues contain higher levels of 27-hydroxycholesterol, a cholesterol metabolite, than normal tissues [37]. Disruption of cholesterol metabolism and its structural involvement in cell membranes, particularly lipid rafts, is a possible therapeutic target in cancer treatment [38]. Statins and other cholesterol-lowering medications have been studied for their chemoprotective properties. Statins block HMG-CoA reductase, a crucial enzyme in the mevalonate pathway, hence lowering cholesterol synthesis [39]. This inhibition also has an effect on downstream products that are necessary for cell growth and signaling. Although clinical trials have had conflicting outcomes, numerous research highlight statins’ anticancer potential due to their pleiotropic properties. These include decreased protein prenylation, decreased tumor cell proliferation and migration, suppression of Ras signaling, and induction of apoptosis via Akt inactivation and mTOR downregulation [40].
While cholesterol is required for regular cellular processes such as steroid and vitamin D3 synthesis, its dysregulation has been linked to cancer development [41]. As a result, targeting cholesterol synthesis and metabolism, notably with statins, may provide intriguing pathways for cancer therapy, despite their impact on normal physiological functions.
Mechanisms of cholesterol deposition in the eye
Cholesterol deposition is involved in several ocular diseases, such as AMD and corneal arcus. Understanding the mechanisms of cholesterol deposition in the eye is crucial to develop effective prevention and treatment strategies. The oxidation of LDL plays a critical role in the deposition of cholesterol in ocular tissues. Oxidized LDL is also taken up by macrophages in the retinal pigment epithelium (RPE), leading to the formation of foam cells and the deposition of cholesterol in Bruch’s membrane, a hallmark feature of AMD [42].
Deposition of cholesterol is caused by dyslipidemia and transport defects in the eye. The ATP-binding cassette transporter A1 (ABCA1), for instance, promotes the efflux of cholesterol from RPE cells. Retinal cholesterol accumulation and drusen development are due to dysfunctional ABCA1, which is a feature of AMD [43, 44]. Additionally, chronic inflammation can lead to cholesterol accumulation in the eye. Inflammatory cytokines, for example, tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), facilitate increased cholesterol uptake and foam cell formation in RPE cells, and hence the occurrence of AMD [45].
Genetic predisposition plays a critical modulatory role in ocular cholesterol deposition by influencing lipid transport, intracellular trafficking, and inflammatory responsiveness within ocular tissues. Variants in apolipoprotein E (APOE), particularly the ε4 allele, alter lipoprotein binding affinity and cholesterol clearance efficiency in the RPE, promoting lipid retention within Bruch’s membrane and accelerating drusen formation [46]. Polymorphisms in ATP-binding cassette transporters, including ABCA1 and ABCG1, impair reverse cholesterol transport and reduce cholesterol efflux to HDL, resulting in intracellular lipid accumulation and heightened susceptibility to oxidative stress [47]. In addition, mutations in lipoprotein lipase (LPL) and cholesteryl ester transfer protein (CETP) genes disrupt systemic lipid handling and indirectly increase ocular cholesterol exposure. Emerging genome-wide association studies further implicate complement-related genes (e.g., CFH and C3) in lipid-rich deposit formation, suggesting a genetic convergence between dysregulated cholesterol metabolism and chronic inflammation in retinal degeneration [48].
Hormonal imbalance also contributes to cholesterol deposition in ocular tissues through its regulatory effects on lipid metabolism, vascular permeability, and cellular cholesterol uptake. Estrogen modulates LDL receptor expression, cholesterol efflux transporters, and antioxidant defenses in the RPE; fluctuations in estrogen levels such as those occurring during menopause are associated with impaired cholesterol clearance and increased lipid deposition in Bruch’s membrane. Glucocorticoids influence hepatic and ocular lipid metabolism by promoting lipogenesis and inhibiting cholesterol efflux pathways, potentially exacerbating retinal lipid accumulation under chronic stress conditions. Thyroid hormone deficiency further alters cholesterol turnover by reducing LDL receptor expression and slowing lipid clearance, thereby increasing circulating cholesterol available for ocular deposition. Collectively, genetic susceptibility and hormonal dysregulation act as permissive factors that amplify cholesterol accumulation in the eye, particularly in aging populations and individuals with metabolic or endocrine disorders [49].
Intake of food can dictate deposition of cholesterol in the eye. High intake of saturated and trans fatty acids increases circulating LDL levels, which can lead to deposition of cholesterol in ocular tissue. High cholesterol diets have been used to induce pathological alteration of the retina, including deposition of cholesterol crystals [50]. A high intake of antioxidants and omega-3 fatty acids can reduce the risk of ocular disease caused by cholesterol [51].
Dysfunction of the blood-brain barrier (BBB) can contribute to cholesterol accumulation in the eye. The choroidal endothelial cells, which maintain the integrity of the BBB in the eye, play a crucial role in regulating cholesterol transport. Disruption of the BBB can lead to an influx of LDL particles into the eye, resulting in cholesterol deposition [52].
Hence, cholesterol deposition in the eye is a complex process influenced by a variety of mechanisms that involve dysregulated lipid metabolism, inflammation, oxidative stress, genetic factors, age-related changes, dietary choices, blood-brain barrier dysfunction, hormonal influences, and excessive cholesterol intake. Understanding these mechanisms is essential for the development of targeted therapies aimed at preventing or mitigating cholesterol-related ocular diseases.
Association between cholesterol and ocular disorders
AMD
Macula, as a highly metabolically demanding tissue, requires a high quantity of cholesterol for optimal function. However, derangement of cholesterol metabolism can potentially lead to adverse effects, such as abnormal lipid deposition and oxidative stress, which play a role in the pathogenesis of several degenerative diseases, including AMD [53]. Various mechanisms have been proposed for the involvement of cholesterol in AMD. Firstly, surplus cholesterol has been implicated in drusen formation, which are extracellular deposits located between Bruch’s membrane and the RPE [54]. Drusen cause disruption of normal cell function and nutrient transport and consequently induce inflammation and oxidative stress in the macula [55]. Secondly, cholesterol impacts the RPE directly, causing it to fail and triggering the formation of cytotoxic metabolites that damage retinal cells. Finally, cholesterol is involved in angiogenesis, the development of pathological new vessels, and its deregulation has been implicated in neovascular AMD, an aggressive variant of the disease [56].
There is growing evidence to support the association between cholesterol and AMD. Epidemiological studies have consistently reported that individuals with high levels of cholesterol, especially LDL cholesterol (“bad” cholesterol), are at greater risk of developing AMD [57]. Furthermore, longitudinal studies have produced evidence of a dose-response gradient for cholesterol and AMD progression and suggest that high levels of cholesterol can worsen the outlook for this disease.
Understanding the role of cholesterol in the pathogenesis of AMD also has important therapeutic implications. Cholesterol-lowering drugs, such as statins, have been used widely in the treatment of cardiovascular disease and may confer additional benefit in reducing the risk of development or progression of AMD. Use of statins has been recently found to have a possible protective effect against AMD, and the drugs may represent a potential future target [58]. However, further research is needed to determine the proper dose and treatment duration. In addition, dietary changes to reduce cholesterol levels, such as following a Mediterranean-type diet rich in fruits, vegetables, whole grains, and healthy fats, have been associated with a decreased risk of AMD.
Retinal vascular diseases
Cholesterol metabolism also plays a role in retinal vascular disorders, including diabetic retinopathy (DR) and retinal vein occlusion (RVO). The aim of this section is to review the relationship between cholesterol and retinal vascular diseases, clarifying the mechanisms, impact, and potential preventive and therapeutic approaches [59].
DR is among the leading causes of blindness worldwide and is associated with high morbidity and mortality in diabetics [60]. Several risk factors that play a fundamental role in the development and progression of DR, including dyslipidemia, particularly elevated cholesterol levels [61]. Chronic hyperglycemia, a feature of diabetes, triggers a cascade of pathophysiological processes, resulting in deranged retinal circulation, oxidative stress, and inflammation [62]. Changes in cholesterol metabolism have been suggested in the pathogenesis of DR [63]. Dyslipidemia, as represented by elevated total cholesterol, LDL cholesterol, and triglyceride levels, has been associated with a high risk of DR progression [64]. The presence of dyslipidemia, particularly hypercholesterolemia, affects the retinal microvasculature.
High cholesterol disrupts endothelial function, vascular permeability, and integrity of the blood-retinal barrier, leading to the development of retinal microaneurysms, hemorrhages, and exudates [65]. Cholesterol can also enhance the generation of oxidative stress with the production of reactive oxygen species (ROS) and subsequent injury to the retinal tissue [66]. Oxidative stress, in turn, triggers inflammatory responses with cytokines and chemokines, which contribute to the development of DR [67]. Pro-inflammatory action mediated via cholesterol can trigger various signaling pathways recruiting and activating immune cells. Inflammatory processes have a central role in the pathogenesis and progression of DR, and cholesterol-mediated inflammation enhances retinal damage and neovascularization [68]. Additionally, dyslipidemia, as caused by hypercholesterolemia, affects retinal endothelial cells, pericytes, and macrophages, leading to retinal neovascularization [69]. Angiogenesis, or new blood vessel formation, is one of the hallmark characteristics of DR. Cholesterol-rich microenvironments promote angiogenesis through multiple molecular mechanisms involving vascular endothelial growth factor (VEGF) and other angiogenic factors. Inhibition of the cholesterol synthesis pathway and regulation of lipid-mediated signaling are promising therapeutic targets for DR [70].
Therapies aiming at cholesterol regulation have also been effective in preclinical and clinical studies. Statins, which are widely used drugs for lowering cholesterol, have also been found to be effective in reducing the risk and development of DR. Novel therapeutic strategies targeting lipid-mediated pathways are also being developed to counteract the harmful effects of cholesterol on the retina [71].
Accumulating evidence shows that the relationship of cholesterol with DR is altered in the presence of other risk factors, such as hypertension, hyperglycemia, and obesity. These risk factors coexist with dyslipidemia and facilitate the development of DR. Clarification of the intricate interaction between cholesterol and these risk factors will culminate in an understanding of the multifactorial etiology of DR and the development of personalized treatment strategies. The relationship between dyslipidemia, especially high cholesterol, and DR is complex and multifactorial. Accumulating evidence suggests that high cholesterol plays a critical role in the pathogenesis and progression of DR, causing microvascular damage, and increasing oxidative stress and inflammation [72]. However, active interventions, including therapeutic interventions of statin therapy and metformin, and lifestyle intervention against dyslipidemia, have been shown to be effective in reducing the risk and severity of DR [73].
RVO is a common vascular retinal disease associated with severe visual loss and blindness [74]. Although hypertension and diabetes are well-proven risk factors, greater importance has been attached to the role of cholesterol in RVO development and progression. Several studies have shown that elevated levels of total cholesterol, LDL cholesterol, and TG are associated with an increased risk of RVO [75]. Cholesterol, particularly LDL, is a cause of atherosclerosis, involving both the large and small vessels. Atherosclerotic changes in the retina cause narrowing of retinal veins, endothelial dysfunction, and thrombosis, especially at the junctions between arteries and veins [76]. These changes are accountable for retinal blood flow impairment and can cause ischemia.
Of the RVO subtypes, branch RVO (BRVO) is the most common and is most frequently associated with systemic vascular risk factors, including high cholesterol levels. BRVO may result from local inflammation, oxidative stress, and thrombogenesis at sites of vascular compression. Central RVO (CRVO), while less common, typically presents with more severe visual impairment and also has been linked to high cholesterol levels [77].
Statins, which are widely used for cholesterol control, have been demonstrated to exert other-than-lipid-lowering effects. Statins improve endothelial function, stabilize atherosclerotic plaques, and reduce inflammation. All these mechanisms might be linked to reduced RVO risk in patients on statins. Clinical trials have shown that the use of statins can improve visual acuity and reduce the risk of recurrence in RVO patients, and therefore it has a possible role in prevention as well as treatment.
Genetic factors are also responsible for susceptibility to RVO in terms of cholesterol metabolism. APOE and LPL gene variants have been found with dyslipidemia and are speculated to have the potential to increase the risk of RVO. Such findings support the idea of individualized RVO management based on genetic profiling and evaluation of risk in individuals. New research is exploring new mechanisms by which cholesterol pathways in the eye are targeted. It has been suggested that the identification of individuals with genetic mutations related to lipid metabolism would provide a rationale for early intervention [78]. This approach might prove beneficial in the prevention of RVO in high-risk individuals.
Cataracts
Cholesterol plays an important role in maintaining the structural stability and transparency of the lens because the lens membrane has the highest cholesterol-to-phospholipid ratio in the human body. This unique lipid structure offers maximal fluidity of the membrane, protein stability, and lens transparency [79]. It is, however, disruptions to cholesterol homeostasis via genetic mutation, systemic hypercholesterolemia, or oxidative damage that make the lens opaque and cause cataract formation [80]. Oxidative stress, in particular, triggers lipid peroxidation, protein aggregation, and crystallin degradation to cause opacification [1]. Defective cholesterol transport mechanisms, including impaired function of ATP-binding cassette (ABC) transporters such as ABCA1, play a crucial role in cataract pathogenesis by dysregulating cholesterol efflux and triggering cell damage to the lens [81].
Hypercholesterolemia and dyslipidemia are increasingly being recognized as systemic risk factors for cataractogenesis. Raised serum cholesterol is associated with increased oxidative damage to the lens, encompassing the production of ROS and alterations in lipid metabolism [82]. This redox environment affects membrane-bound proteins and incapacitates lens epithelial cell function, favoring cortical and nuclear cataractogenesis. In addition, metabolic disorders such as diabetes and metabolic syndrome, which are often associated with dyslipidemia, promote lens damage through advanced glycation end-products (AGEs) and oxidative mechanisms. Clinical research and animal models have shown that lipid-lowering drugs, including statins, in addition to their ability to reduce systemic cholesterol levels, may also be protective against cataract formation, but the long-term effect of these interventions on lens health remains an area of current investigation [83, 84].
Excess cholesterol contributes to cataractogenesis by disrupting lens membrane architecture and protein homeostasis, both of which are essential for maintaining lens transparency. The lens fiber cell membrane is uniquely enriched in cholesterol, and pathological increases further enhance membrane rigidity, reducing fluidity and impairing the function of critical membrane proteins such as aquaporin-0 and connexins that regulate water transport and intercellular communication [1, 2]. These alterations compromise ionic and osmotic balance within the lens, leading to fiber cell swelling and increased light scattering. In addition, cholesterol-rich membrane domains promote the formation of cholesterol crystalline phases, which destabilize membrane organization and interfere with lens fiber cell packing [3]. Oxidative stress further exacerbates this process through the generation of cholesterol oxidation products (oxysterols), which induce ER stress, mitochondrial dysfunction, and apoptotic signaling in lens epithelial cells [4, 5]. Cholesterol-driven changes in lipid raft composition also disrupt protective signaling pathways and accelerate crystallin protein misfolding, aggregation, and insolubilization – hallmark molecular events in age-related nuclear cataract formation [6]. Moreover, excessive cholesterol accumulation increases susceptibility of lens membranes to ultraviolet-induced lipid peroxidation, amplifying oxidative damage and accelerating cataract progression with aging [5, 7].
Recent studies have highlighted the critical role of cholesterol homeostasis in maintaining lens transparency and preventing cataract formation. SREBP2-mediated cholesterol biosynthesis has been identified as a key pathway regulating lens membrane composition and protein stability, with disruption leading to protein aggregation and opacification of the lens [85]. In human cataractous lenses, altered expression of enzymes involved in cholesterol metabolism, such as lanosterol synthase and 7-dehydrocholesterol reductase, has been observed, indicating that impaired local sterol synthesis contributes to age-related lens degeneration [86]. Genetic studies further support this link; variants in HMGCR and other cholesterol-lowering genes are associated with an increased risk of cataract formation, suggesting that lifelong alterations in systemic cholesterol metabolism may influence lens homeostasis and susceptibility to opacification. At the molecular level, cholesterol depletion or imbalance in the lens membrane disrupts lipid rafts and α-crystallin interactions, destabilizing protein conformation and promoting aggregation. Oxidative modifications of cholesterol and other membrane lipids exacerbate these effects by increasing ROS-mediated damage, impairing lens protein repair mechanisms, and ultimately triggering cataractogenesis [87]. Experimental models corroborate these findings, showing that pharmacologic or genetic inhibition of cholesterol biosynthesis in the lens induces cataract formation, emphasizing the necessity of maintaining local cholesterol levels for lens clarity [88].
Additionally, hormonal imbalances – particularly changes in corticosteroids, estrogen, and thyroid hormones – can alter systemic and local cholesterol metabolism, indirectly affecting lens membrane composition and susceptibility to oxidative stress. These hormonal influences may modify the expression of key cholesterol biosynthetic enzymes and antioxidant defense pathways, further contributing to cataract formation in both age-related and secondary cataracts. Collectively, these findings underscore a multifactorial pathophysiological mechanism, where genetic predisposition, disturbed cholesterol metabolism, oxidative damage, and hormonal regulation converge to compromise lens transparency and promote cataractogenesis.
Glaucoma
Cholesterol metabolism has emerged as a critical factor in the pathophysiology of glaucoma, a progressive optic neuropathy characterized by retinal ganglion cell (RGC) loss and optic nerve damage [89]. Cholesterol homeostasis in the retina and optic nerve is tightly regulated to ensure proper neuronal function and membrane integrity. Dysregulation of cholesterol transport and metabolism, mediated by aberrant activity of LDL, HDL, and ATP-binding cassette (ABC) transporters such as ABCA1 and ABCG1, has been implicated in glaucomatous damage [90]. Excess cholesterol accumulation in RGCs and surrounding glial cells promotes oxidative stress, inflammation, and mitochondrial dysfunction, ultimately exacerbating neuronal apoptosis. These findings suggest a complex interplay between lipid metabolism and glaucomatous neurodegeneration.
Epidemiological studies have highlighted an association between systemic dyslipidemia and an increased risk of glaucoma, particularly primary open-angle glaucoma (POAG) [91]. Elevated serum cholesterol levels, alongside altered LDL-to-HDL ratios, have been linked to impaired aqueous humor outflow and elevated intraocular pressure (IOP), a key modifiable risk factor for glaucoma [92]. Cholesterol accumulation in the trabecular meshwork disrupts cellular function and extracellular matrix remodeling, leading to reduced drainage and elevated IOP [93]. Moreover, systemic lipid abnormalities may influence optic nerve head perfusion and vascular health, further contributing to glaucomatous damage. Statins, widely prescribed for hypercholesterolemia, have shown potential neuroprotective effects in glaucoma patients by improving endothelial function, reducing oxidative stress, and modulating lipid metabolism, although their precise role in glaucoma management remains under investigation.
Molecular pathways implicated in cholesterol-related ocular diseases
The pathogenesis of cholesterol-related ocular diseases is based on multifaceted molecular signal transduction pathways that control lipid metabolism, inflammation, angiogenesis, oxidative stress, and neurodegeneration. Understanding these pathways provides insights to facilitate the discovery of novel therapeutics as well as optimization of existing treatments.
Mevalonate pathway and SREBP-2 regulation
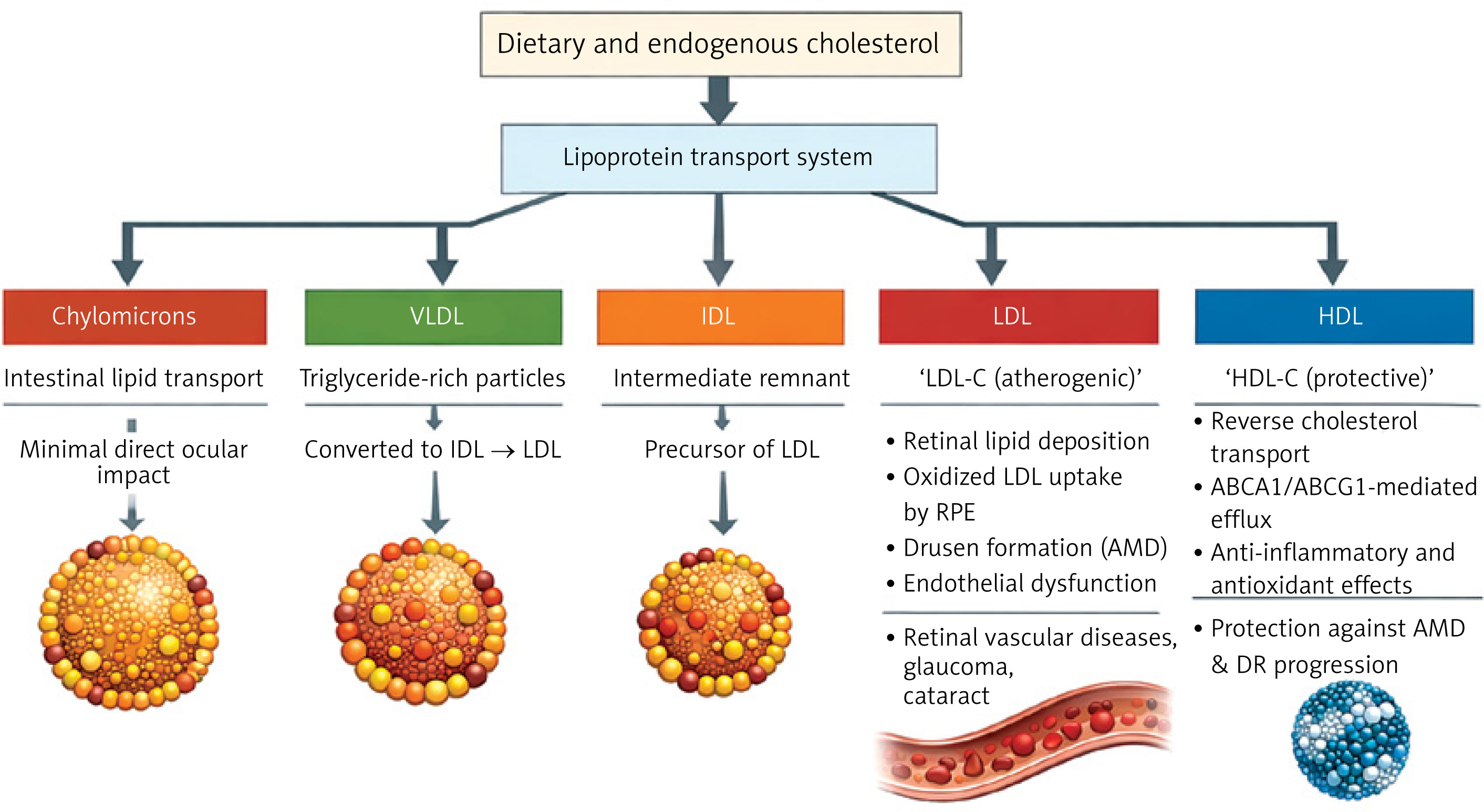
Cholesterol biosynthesis begins with acetyl-CoA, which is converted to mevalonate by the rate-limiting enzyme 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR). The mevalonate route involves multiple intermediates, including squalene and lanosterol, before producing cholesterol and necessary isoprenoids for cellular activity [94]. The transcriptional regulator of this pathway is sterol regulatory element-binding protein 2 (SREBP2). SREBP2 increases the expression of genes such as HMGCR and LDL receptor genes, promoting cholesterol synthesis and absorption. It is first produced as an inactive precursor embedded in the ER membrane. When intracellular cholesterol levels are low, SREBP2 is transferred to the Golgi apparatus and cleaved to release the active domain. This domain then penetrates the nucleus and activates target gene expression [21].
However, under high cholesterol conditions, SREBP2 activation is suppressed through a feedback mechanism involving SCAP and insulin-induced gene (INSIG) proteins. SCAP binds to SREBP2 and, in the presence of sufficient cholesterol, interacts with INSIGs in the ER, anchoring the complex and preventing its transport to the Golgi [95, 96]. This regulatory system maintains cholesterol homeostasis and prevents overproduction. Disruption of the cholesterol manufacturing pathway can cause aberrant cholesterol accumulation in tissues, which contributes to illnesses including AMD. Dysregulation of the mevalonate system, in particular, causes cholesterol deposits in ocular tissues, particularly the RPE and Bruch’s membrane, which promotes the production of drusen, a hallmark of early AMD [57]. Drusen are yellowish-white extracellular deposits seen beneath the RPE. They are made up of lipids, amyloid, complement proteins, crystallins, and other cell detritus. The presence of drusen is one of the first and most distinguishing pathogenic characteristics of AMD. Their size and number are strongly associated with the probability of development of early to severe stages of the disease. Larger and more frequent drusen increases the likelihood of retinal degeneration [97].
Given the importance of cholesterol in drusen formation, the mevalonate pathway, which regulates cholesterol production, is regarded as a key therapeutic target (Figure 2). Statins, which inhibit HMG-CoA reductase (HMGCR) in this pathway, are routinely used to reduce cholesterol levels in cardiovascular and metabolic disorders and have also been investigated as potential AMD therapies. Statins, which reduce intracellular cholesterol synthesis, may help restrict drusen growth and decrease AMD progression, while clinical effects are still being investigated.
ABCA1/ABCG1-mediated reverse cholesterol transport
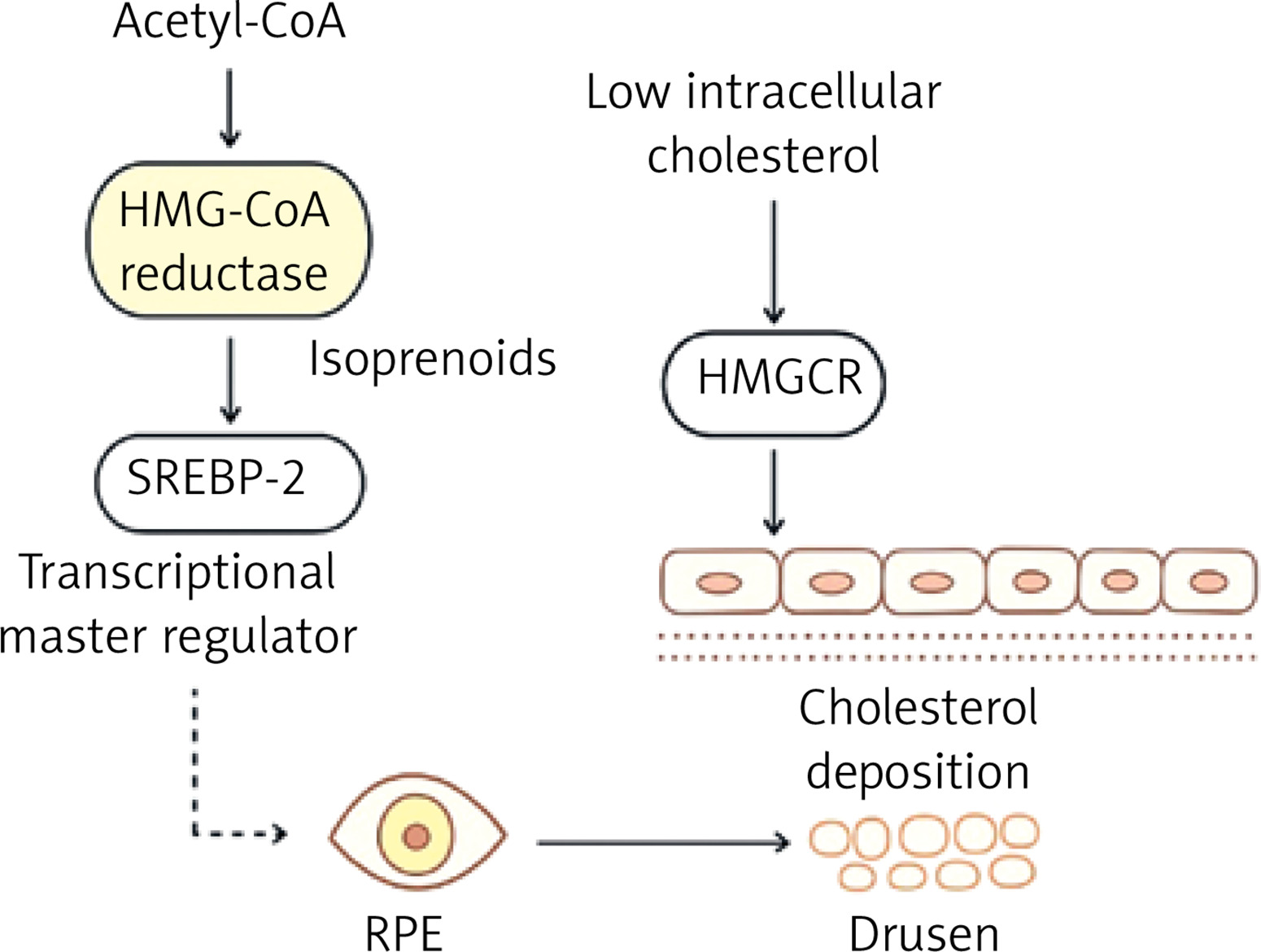
Cholesterol efflux from retinal pigment epithelial (RPE) and glial cells is essential for maintaining lipid homeostasis in the retina. This efflux is mediated by ATP-binding cassette (ABC) transporters, including ABCA1 and ABCG1, which promote the transfer of cholesterol to apolipoprotein acceptors such as ApoA1 and HDL [98]. Dysfunction of these transporters, particularly ABCA1, has been associated with pathological cholesterol buildup, oxidative stress, and retinal damage [99]. ABCA1 is widely expressed in organs including the liver, intestines, adipose tissue, and macrophages, where it controls cholesterol and phospholipid efflux as well as cellular signaling [100].
The liver X receptor (LXR), a ligand-activated nuclear receptor that responds to intracellular oxysterols, which accumulate when cholesterol levels are high, regulates transcription of both ABCA1 and ABCG1 [101]. LXR is a critical regulator of the RCT pathway. When activated, LXR forms a heterodimer with the retinoid X receptor (RXR), which binds to certain DNA response regions and promotes transcription of target genes such as ABCA1, ABCG1, SREBP-1c, and ApoE [102]. ABCG1, albeit less explored than ABCA1, is critical in intracellular sterol distribution and export to mature HDL particles [100]. Unlike ABCA1, which enables cholesterol transfer to lipid-poor ApoA1, ABCG1 promotes cholesterol efflux to lipid-rich HDL, which helps to maintain cholesterol balance. ABCG1 is mostly present in intracellular compartments, and its function appears to be cell type specific [103]. ABCG1 dysregulation has been linked to a number of diseases, including atherosclerosis, Alzheimer’s disease, and metabolic disorders [104].
LXR agonists have shown therapeutic promise in preclinical models of retinal degeneration by increasing cholesterol efflux and decreasing inflammation. These medicines increase the expression of ABCA1 and ABCG1, which reduce cholesterol accumulation in RPE cells and modulate inflammatory pathways, including inhibiting NF-κB signaling [105]. LXRα and LXRβ, the two isoforms of LXR, are distributed differently throughout tissues, with LXRβ being more widely expressed. Overall, the LXR-ABCG1 axis is critical in regulating cholesterol levels in the retina and other tissues. Its activation could be a therapeutic target for disorders characterized by lipid imbalance and persistent inflammation [101] (Figure 3).
Figure 3
Regulation of retinal cholesterol homeostasis via liver X receptor (LXR)-ABCA1/ABCG1 signaling. In RPE cells and glial cells, cholesterol efflux is mediated by the ATP-binding cassette transporters ABCA1 and ABCG1. Activation of LXR upregulates ABCA1 expression, thereby enhancing cellular cholesterol efflux to extracellular apolipoprotein acceptors. This process facilitates reverse cholesterol transport and prevents intracellular cholesterol accumulation. Therapeutic LXR agonists may augment this pathway, promoting cholesterol clearance and contributing to the maintenance of retinal lipid homeostasis
VEGF and angiogenic pathways
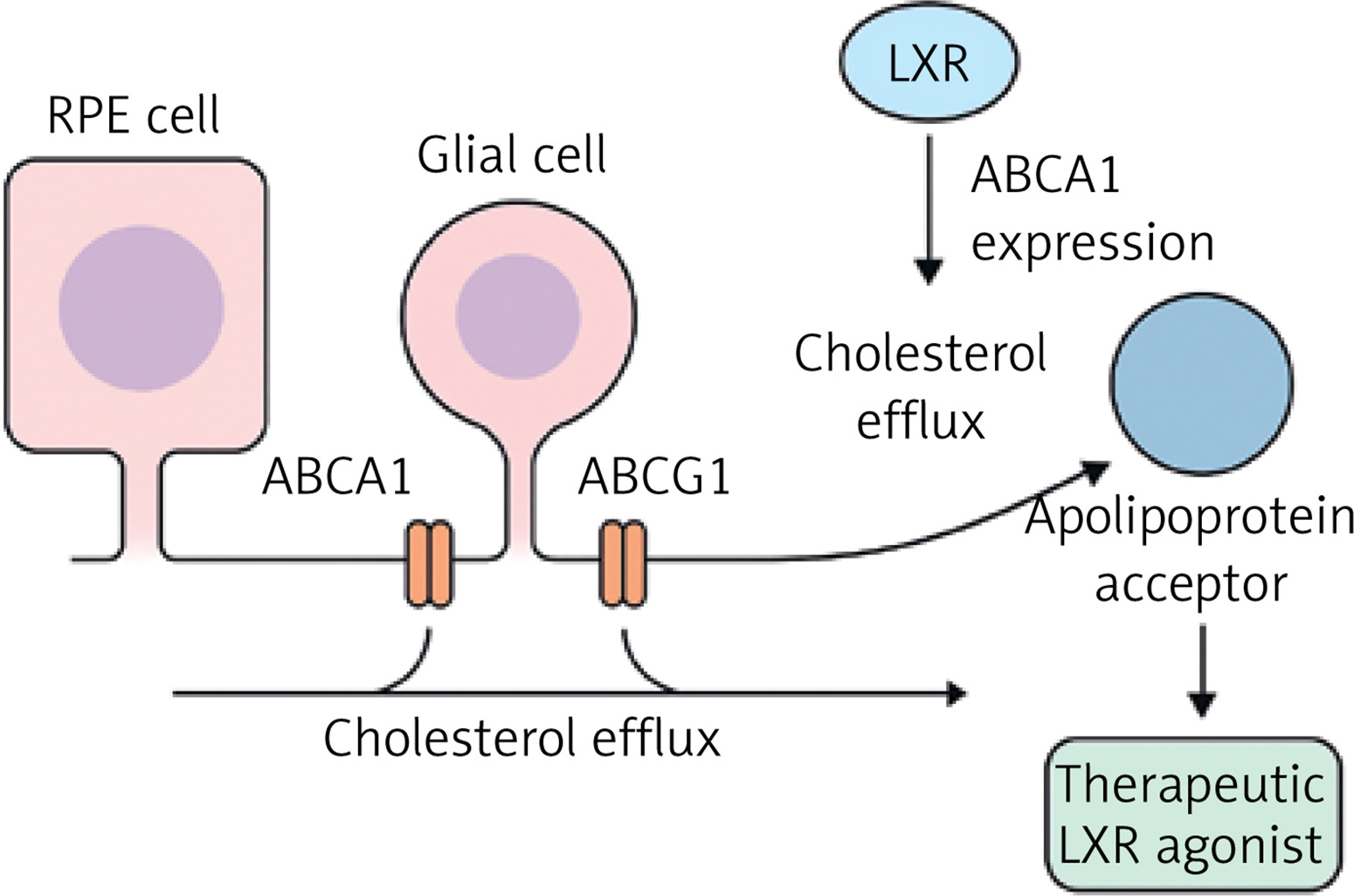
VEGF is a crucial regulator of angiogenesis, or the development of new blood vessels, and is involved in both normal physiological processes and pathological disorders such cancer, preeclampsia, and ophthalmic disorders. The VEGF family consists of VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, VEGF-F, placental growth factor (PlGF), and endocrine gland-derived VEGF (EG-VEGF), all of which have a cystine knot structural motif. VEGF proteins form dimers and bind to certain tyrosine kinase receptors (VEGFR-1, VEGFR-2, and VEGFR-3), triggering intracellular signaling cascades that control vascular permeability, endothelial cell migration, and lymphangiogenesis [106].
VEGF’s critical function in pathological angiogenesis is well recognized in AMD and DR, the two primary causes of visual loss. Chronic metabolic stress, such as dysregulated lipid metabolism, oxidative stress, and hypoxia, promotes the overexpression of VEGF, particularly VEGF-A. This causes aberrant neovascularisation, increased vascular permeability, and the collapse of the blood-retinal barrier [107]. In wet AMD, high VEGF causes the development of fragile, leaky blood vessels beneath the retina, resulting in fluid accumulation and vision impairment [108]. Elevated VEGF causes capillary leakage in diabetic macular edema (DME), a consequence of DR, which is exacerbated by the failure of Müller glial cells that usually regulate fluid balance in the retina [109].
The VEGF/VEGFR2 signaling pathway stimulates downstream mediators such as PI3K/Akt and MAPK/ERK, which promote endothelial cell survival, proliferation, and migration [110]. Anti-VEGF treatments, such as ranibizumab and aflibercept, target this system to minimize pathological angiogenesis and vascular leakage in ocular disorders [111]. In contrast, VEGF does not have a direct role in dry AMD, which is characterized by drusen buildup and RPE dysfunction [108]. However, the chronic hypoxic and inflammatory milieu in dry AMD may indirectly upregulate VEGF as a compensatory response to reduced choroidal perfusion [112].
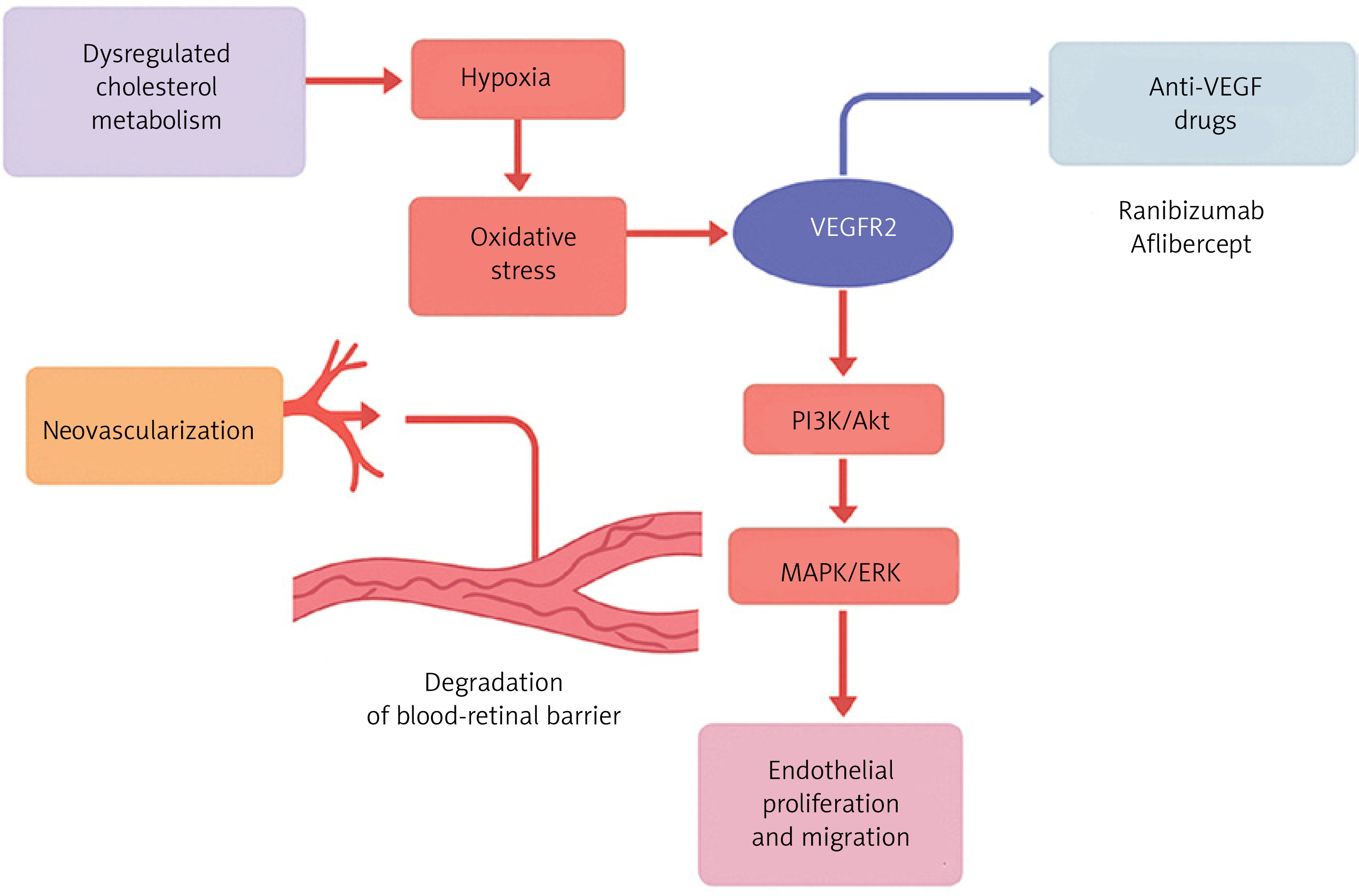
Aside from the eye, VEGF plays an important function in diabetic nephropathy, where chronic hyperglycemia and metabolic stress cause hypoxic conditions in the renal milieu [113]. Hypoxia in the kidney increases VEGF expression in renal cells, particularly mesangial cells and podocytes, as a compensatory reaction to improve oxygen delivery through angiogenesis. However, chronic hypoxia-induced VEGF overexpression breaks the glomerular filtration barrier by increasing vascular permeability, resulting in proteinuria and glomerular injury. The presence of VEGF receptors (VEGFRs) on kidney cells mediates the detrimental effects [114]. This mechanism is similar to the retinal vascular alterations observed in diabetic retinopathy, where hypoxia-induced VEGF overexpression causes aberrant neovascularisation and vascular leakage. Thus, hypoxia is a critical upstream driver for VEGF dysregulation in both diabetic nephropathy and retinopathy, connecting poor oxygen delivery to pathological angiogenesis and vascular dysfunction in diabetes [115] (Figure 4).
Figure 4
Proposed mechanistic pathway linking dysregulated cholesterol metabolism to retinal neovascularization and barrier breakdown via VEGF signaling. Dysregulated cholesterol metabolism induces tissue hypoxia and oxidative stress, leading to activation of vascular endothelial growth factor receptor-2 (VEGFR2). Subsequent stimulation of downstream PI3K/Akt and MAPK/ERK signaling cascades promotes endothelial cell proliferation and migration, contributing to pathological neovascularization and degradation of the blood–retinal barrier. Anti-VEGF therapies, including ranibizumab and aflibercept, act by inhibiting VEGFR2-mediated signaling, thereby attenuating angiogenic responses and vascular permeability
Other pathways
Oxidative stress and NRF2 pathway
Cholesterol buildup in retinal cells greatly boosts the formation of ROS, particularly within mitochondria, resulting in oxidative stress. This oxidative stress plays a vital role in the development of eye disorders such as glaucoma, DR, and AMD. The retina’s high oxygen consumption and continual exposure to external stimuli such as ultraviolet radiation make it particularly susceptible to ROS-induced damage. Certain substances or mechanisms in the retina also have cytoprotective effects, which serve to reduce oxidative damage [116].
The Keap1-NRF2-ARE pathway is an important cellular defense mechanism against oxidative stress. Under normal settings, Kelch-like ECH-associated protein 1 (Keap1) binds to NRF2 in the cytoplasm, causing it to be ubiquitinated and degraded, resulting in low basal NRF2 levels [117]. Cysteine residues on Keap1 are changed in response to oxidative stress, preventing it from tagging NRF2 for destruction. This permits NRF2 to detach from Keap1, move into the nucleus, and bind to antioxidant response elements (AREs), boosting the production of antioxidant and cytoprotective genes such heme oxygenase-1 (HO-1) and glutathione peroxidase. HO-1 is an essential phase II detoxifying enzyme that is modestly expressed under normal conditions but increases considerably during oxidative stress to protect retinal cells from harm [118]. Studies have identified NRF2 as a master regulator of antioxidant responses and a prospective therapeutic target for a variety of illnesses. For example, 24-hydroxycholesterol (24-OHC), a cholesterol metabolite, activates the deacetylase SIRT1, which increases NRF2 expression and has neuroprotective benefits [119]. Dysregulation of the NRF2 pathway causes oxidative damage in retinal photoreceptors and lens cells, which contributes to AMD development and cataract formation [120].
Therapies such as the Age-Related Eye Disease Studies (AREDS2) antioxidant formulation seek to indirectly stimulate this system, providing protection against oxidative damage in age-related eye disorders [121]. Furthermore, NRF2 expression in glaucoma trabecular meshwork cells is lower than in healthy cells. Nonetheless, experimental overexpression of NRF2 has been found to accelerate cell proliferation and suppress apoptosis in these cells [122]. Furthermore, several studies have shown that increasing NRF2 signaling lowers ROS generation, hence protecting retinal ganglion cells, retinal epithelial cells, and lens cells from oxidative stress-induced damage [123, 124]. In summary, cholesterol-induced ROS formation causes oxidative stress in the retina, which is reduced by activating the Keap1-NRF2-ARE pathway. Proper control of this system is critical for retinal health and offers intriguing therapeutic options for visual illnesses caused by oxidative damage.
Inflammatory pathways: STING and NF-κB
The cyclic GMP-AMP synthase (cGAS)–stimulator of interferon genes STING pathway is an important innate immune signaling mechanism that identifies cytosolic DNA and induces antiviral and inflammatory responses. cGAS is a DNA sensor that detects double-stranded DNA (dsDNA) in the cytosol, which can come from either endogenous sources (such as damaged mitochondria, genomic instability, or retrotransposition) or external sources, such as pathogen DNA (viruses, bacteria), or dead cells [109]. When cGAS recognizes DNA, it catalyzes the conversion of ATP and GTP into a second messenger, cyclic GMP-AMP (cGAMP). cGAMP then binds and activates STING, a transmembrane adaptor protein found in the ER. Once triggered, STING moves from the ER to the Golgi apparatus via the ER-Golgi intermediate compartment (ERGIC). At the Golgi, active STING recruits and interacts with phosphorylated TBK1 and IKK. This causes phosphorylation of IRF3 and IκBα. Phosphorylated IRF3 moves into the nucleus and activates the transcription of type I interferon (IFN-I) genes, which are essential for antiviral defense [125]. Phosphorylation of IκBα stimulates NF-κB, which regulates the expression of pro-inflammatory cytokines [126]. The cGAS-STING pathway connects cytosolic DNA recognition to the activation of both interferon-mediated antiviral immunity and inflammatory signaling, playing critical roles in infection responses, tumor immunity, and autoimmune disorders.
Chronic inflammation is a feature of cholesterol-induced retinal disease. The NF-κB signaling pathway is activated by cytokines such as TNF-α and IL-1β, commonly caused by cholesterol crystal formation and the uptake of oxidized LDL [127]. NF-κB translocation leads to expression of pro-inflammatory mediators involved in retinal damage and blood vessel leakage. The STING pathway, activated by intracellular cholesterol crystals, contributes to retinal endothelial inflammation and microvascular damage [69]. Although not yet targeted therapeutically in ophthalmology, these pathways are being explored as targets for potential intervention.
PPAR-α pathway in lipid and vascular homeostasis
Peroxisome proliferator-activated receptor α (PPAR-α) is a nuclear receptor that regulates lipid metabolism and fatty acid oxidation. It is a member of the nuclear hormone receptor superfamily, which includes both classical hormone receptors (e.g., glucocorticoid and thyroid hormone receptors) and metabolic sensors such as PPARs and liver X receptors. PPAR-α is highly expressed in metabolically active tissues with enhanced fatty acid β-oxidation, such as liver, heart, kidney, skeletal muscle, brown adipose tissue, and retina. PPAR-α is found in retinal vascular cells responsible for choroidal neovascularization (CNV) [128]. Clinically, PPAR-α is the primary target of fibrate drugs (including fenofibrate, gemfibrozil, and clofibrate), which are used to lower serum TG and cholesterol [129]. Apart from lipid regulation, PPAR-α activation has been shown to have anti-inflammatory and anti-angiogenic effects, particularly in retinal diseases. Fenofibrate, a PPAR-α agonist, has demonstrated efficacy in reducing retinal inflammation and neovascularization, key features of DR [130]. Clinical trials have shown that fenofibrate can reverse retinal endothelial dysfunction and inhibit VEGFR2 expression, a key mediator in pathological angiogenesis [131, 132]. Varet et al. further confirmed the anti-angiogenic effect of fenofibrate both in vitro and in vivo, highlighting the therapeutic potential of PPAR-α activation in retinal vascular diseases [133].
Emerging therapies in cholesterol-linked eye diseases
The therapeutic avenue of cholesterol-associated ocular diseases demands a focus on the management of disorders comprising disrupted lipid metabolism, oxidative injury, and inflammatory responses. Interventions must be targeted to avoid retinal deterioration, vascular breakdown, and neuronal injury, which are all dominant characteristics of conditions such as AMD, DR, cataracts, and glaucoma. One of the most well-documented pharmacological classes used to treat cholesterol-induced ocular disease is the statins – such as atorvastatin, simvastatin, and rosuvastatin – all of which are commercially available and approved [134]. Statins exert their effect by inhibiting HMG-CoA reductase in the mevalonate pathway, thereby reducing endogenous cholesterol synthesis. Apart from their lipid-lowering effect, statins have been reported to improve endothelial function, reduce oxidative stress, and have anti-inflammatory effects. These pleiotropic effects contribute to their protective role in AMD, DR, and RVO. In research studies, statins have been suggested to retard AMD progression and reduce retinal vascular events.
Fenofibrate, also an approved drug, targets PPAR-α. It controls lipid metabolism and reduces TG and inhibits vascular inflammation. Fenofibrate has shown benefit in large trials such as the FIELD and ACCORD-EYE studies by retarding DR progression [135]. It is particularly useful in reducing retinal leakage and microvascular complications caused by dyslipidemia in patients with diabetes.
Anti-vascular endothelial growth factor (anti-VEGF) agents are the treatment of choice for neovascular AMD and diabetic macular edema, where pathological angiogenesis is a secondary effect of hypoxia and deposition of lipid. Of the commercially available agents, these include ranibizumab (Lucentis), aflibercept (Eylea), and bevacizumab (Avastin) (off-label) [136]. Each of these drugs inhibits VEGF-A, thereby reducing neovascularization, vascular leak, and retinal edema. Although their primary mechanism is not against cholesterol itself, these drugs are highly important in controlling downstream consequences of lipid-mediated retinal pathology.
From the antioxidant perspective, the AREDS and AREDS2 formulas of vitamin C, vitamin E, zinc, copper, lutein, and zeaxanthin are recommended supplements that have been shown to be effective in inhibiting progression to late AMD [137]. These vitamins reverse ROS and stabilize cell structures within the retina, particularly in high oxidative stress conditions caused by lipid peroxidation. Although tocotrienols, a very active derivative of vitamin E possessing strong antioxidant activity, had shown preclinical potential in preventing lipid-induced retinal degeneration, they are not yet ophthalmic drugs on the market and hence are not regarded as commercially available treatments. Together, these approved therapies target key pathways including the HMG-CoA reductase–mevalonate pathway, PPAR-α-controlled lipid homeostasis, and VEGF-induced angiogenesis, along with antioxidant defense to reduce oxidative damage in lens and retina.
New gene therapies are being developed to express anti-VEGF proteins, potentially reducing the need for frequent injections in AMD patients [138]. Biodegradable nanoparticles are being explored for targeted delivery of therapies to ocular tissues, enhancing treatment efficacy and reducing side effects [139]. While these emerging therapies show promise, challenges remain, such as the need for further clinical validation and the complexity of ocular drug delivery systems. The ongoing research into lipid metabolism and its implications for ocular health continues to evolve, indicating a dynamic landscape in the treatment of cholesterol-linked eye diseases. This multifaceted approach generally prevents cholesterol-induced damage to ocular tissue and is the foundation for multimodal treatment regimens (Table I).
Table I
Emerging therapies in cholesterol-linked eye diseases
Key highlights of this review
Integrative lipid-centered perspective:
This review unifies current evidence linking cholesterol dysregulation to major ocular disorders including AMD, retinal vascular diseases, cataracts, and glaucoma within a single mechanistic framework.
Beyond total cholesterol: lipoprotein-specific insights:
The manuscript distinguishes the differential roles of LDL-C, HDL-C, and hypertriglyceridemia in ocular pathology, providing a more precise and clinically relevant interpretation than prior cholesterol-focused reviews.
Comprehensive molecular pathway integration:
Key cholesterol-regulated pathways, including the mevalonate SREBP-2 axis, ABCA1/ABCG1-mediated reverse cholesterol transport, VEGF-driven angiogenesis, NRF2 antioxidant signaling, STING-NF-κB inflammation, and PPAR-α regulation, are systematically integrated.
Translational relevance to current and emerging therapies:
The review critically evaluates approved lipid-modulating and downstream therapies (statins, fenofibrate, anti-VEGF agents, AREDS supplements) alongside emerging approaches such as gene therapy and nanoparticle-based drug delivery.
Forward-looking precision medicine framework:
By incorporating genetic susceptibility, metabolic modifiers, and aging-related changes, this review highlights cholesterol metabolism as a modifiable target for personalized prevention and treatment of vision-threatening ocular diseases.
Conclusions and future perspectives
This review consolidates growing evidence that cholesterol dysregulation is not merely a systemic metabolic abnormality but a clinically relevant contributor to multiple vision-threatening ocular diseases, including AMSD, retinal vascular diseases, cataracts, and glaucoma. By integrating lipoprotein-specific effects, ocular tissue-specific mechanisms, and key molecular pathways, this work provides a clinically meaningful framework that extends beyond traditional cardiovascular interpretations of cholesterol.
From a real-world clinical perspective, the findings of this review support a more integrated, lipid-aware approach to ophthalmic care. Dyslipidemia – particularly elevated LDL-C, reduced HDL-C, and hypertriglyceridemia – should be recognized as modifiable risk factors that may influence disease onset, progression, and therapeutic response in ocular disorders. Routine lipid profiling in patients with AMD, diabetic retinopathy, retinal vein occlusion, cataracts, and glaucoma may therefore offer added value for risk stratification and long-term disease management.
Importantly, widely used systemic therapies already embedded in clinical practice such as statins and fenofibrate may confer ocular benefits beyond their cardiovascular indications through pleiotropic effects on inflammation, oxidative stress, endothelial function, and lipid homeostasis. While anti-VEGF therapies remain the cornerstone for neovascular retinal diseases, this review highlights that lipid modulation addresses upstream drivers of pathology and may complement existing treatment strategies, particularly in chronic and progressive disease states. Nutritional interventions and antioxidant supplementation, as exemplified by AREDS formulations, further reinforce the translational relevance of targeting cholesterol-related oxidative injury in routine care.
The evidence presented underscores the need for closer collaboration between ophthalmologists, endocrinologists, cardiologists, and primary care physicians. Integrated management of systemic metabolic health including lipid control, glycemic regulation, and blood pressure optimization may yield meaningful benefits for ocular outcomes. This approach aligns with preventive medicine principles and supports earlier intervention before irreversible retinal or lenticular damage occurs.
Looking forward, several priorities emerge. Longitudinal clinical studies are needed to define lipid thresholds that are specifically predictive of ocular disease risk and progression, rather than extrapolated from cardiovascular endpoints. Precision medicine approaches incorporating genetic variants (such as APOE, ABCA1, and CETP), lipidomic profiling, and advanced retinal imaging may enable individualized risk prediction and targeted therapy. At the therapeutic level, emerging strategies such as LXR modulators, gene-based anti-VEGF delivery systems, nanoparticle-mediated lipid targeting, and NRF2- or PPAR-α-directed interventions hold promise for addressing cholesterol-driven ocular pathology at a mechanistic level. Future trials should evaluate these approaches not only for anatomical outcomes but also for functional vision preservation and long-term safety.
In summary, cholesterol metabolism represents a clinically actionable and biologically central axis in ocular disease pathogenesis. By reframing cholesterol as a shared determinant of systemic and ocular health, this review supports a shift toward earlier, integrated, and mechanism-based interventions. Such an approach has the potential to improve visual outcomes, reduce disease burden, and advance personalized care for patients at risk of cholesterol-linked eye diseases.