Introduction
Primary hepatic cancer is one of the most common malignancies in the world. The incidence rate of this disease is fifth among malignant tumours, and the mortality rate is third [1, 2]. Although the treatment techniques for hepatic cancer, such as surgical resection, liver transplantation, radiotherapy, and chemotherapy, have been improved, the 5-year survival rate of hepatic cancer patients has not significantly improved [3]. Studies have shown that the development and progression of hepatic cancer are accompanied by abnormalities in many molecular and signalling pathways. However, the mechanisms underlying these aberrant molecules and signalling pathways are still unclear [4, 5]. Therefore, it is of particular importance to study the mechanism of hepatic cancer development and to take specific molecules as markers and therapeutic targets at the molecular level to improve the early diagnosis and prognosis of hepatic cancer patients. Long non-coding RNA (lncRNA) has been a focus of research in recent years. Abnormal expression of lncRNA has been found in a variety of tumours, and these lncRNAs are mainly involved in tumour formation, invasion, metastasis, and recurrence, suggesting that lncRNA acts as an oncogene or tumour suppressor in the development and progression of tumours. Related studies showed that lncRNA and HOTAIR were highly expressed in liver cancer tissues and that the proliferation, invasion, and metastasis ability of hepatic cancer cells after HOTAIR were significantly reduced [6]. The expression of MEG3 was low in hepatic cancer tissues and cell lines. After overexpression of MEG3, the growth of hepatic cancer cells was inhibited and the rate of apoptosis was increased, which indicated that MEG3 could inhibit tumour cells in hepatic cancer [7]. In this study, which used bioinformatics analysis of the gene-chip data GSE33113 public platform, we found that the expression of LINC00152 in colon cancer tissue increased significantly by its in situ hybridisation (ISH) detection in 30 pairs of hepatic cancer and adjacent tissues, and explained the effects and mechanisms of LINC00152 in the development and occurrence of hepatocellular carcinoma.
Material and methods
Sample and collecting
Thirty pairs of cancer and adjacent normal tissues were collected from hepatic cancer patients who were treated in the Third Affiliated Hospital of Sun Yat-Sen University. No patients had been treated with radiotherapy, chemotherapy, immunotherapy, or interventional therapy, nor had they undergone liver transplantation. The specimens were prepared by 4% paraformaldehyde (0.1 mol/l, PBS, containing 0.1% diethylpyrocarbonate, DEPC, pH = 7.4) fixation followed by conventional dehydration. Transparent and paraffin-embedded 4-µm thick continuous slices were subsequently prepared.
In situ hybridization (ISH)
ISH kits MK10098-h and DAB colour kits were purchased from Wuhan Dr & Co. Ltd., and Harris hematoxylin was purchased from Beijing Sino fir Jinqiao Corporation, DEPC, purchased from Sigma Corporation (United States). Oligonucleotide probes were designed and synthesised by Dr Wuhan & Co., and the 3’ end was labelled with digoxin. We referred to the reagent kit instructions and made a number of improvements: paraffin section, regular dewaxing and hydration, PBS/DEPC instead of DEPC, washing for 5 min, 3 times, and inactivation of endogenous peroxidase by 3% H2O2 at room temperature for 10 min. DEPC water immersion was performed three times, followed by addition of fresh pepsin diluted with 3% citric acid in a volume of 30 µl, digestion for 20 min at 37°C, and washing by PBS/DEPC for 5 min, three times. Next, slices were treated with DEPC and fixed with 1% paraformaldehyde (0.1 mol/l PBS preparation, pH = 7.4) at room temperature for 10 min, before DEPC was rinsed in water three times and each slice was dropped in a pre-hybridisation solution of 20 l. Slices were maintained at a constant temperature of 37°C overnight (16–18 h). After hybridisation, 2 × SSC, 0.5 × SSC and 0.2 × SSC gradient were washed at 37°C to wash away the excess probes. The slice was dripped with a closed liquid of 50 µl, closed at 37°C for 30 min before 50 µl of biotinylated mouse anti-human digoxin antibody (i.e., type) was added and the slices incubated at 37°C for 120 min. Slices were washed in PBS for 5 min, four times; 50 µl of streptavidin-biotin complex was added; and the slices were incubated at 37°C for 30 min. Slices were washed in PBS for 5 min, three times; 50 µl streptavidin-biotin complex was added; and slices were incubated at 37°C for 30 min, before being washed in PBS for 5 min, three times, stained with DAB for 5 min, and stained with haemotoxylin. After dehydration, transparent, covering. Instead of hybridisation, the hybridisation solution without probes was used as a negative control.
Immunohistochemistry (IHC)
Monoclonal antibodies kits for EGFR, PI3K, AKT, P21, and MMP-2/9 SP were purchased from the ZYMED Company. The slices were dehydrated with xylene solution, and then rewatered in 95% ethanol, 85% ethanol and 75% ethanol, washed with distilled water, put into 1×citric acid disodium hydrogen phosphate buffer solution for antigen microwave repair for 2 min after the slices were naturally cooled, they were sealed in 3% H2O2 solution for 15 min, and then soaked in PBS to wash the slices. EGFR, PI3K, Akt, p21 and MMP-2/9 antibodies diluted at 1 : 500 were added. PBS was used as the negative control instead of the first antibody. The second antibody coupled with biotin was added and incubated at 37°C for 30 min. After PBS washing, DAB reagent kit (Beijing solebao Technology Co., Ltd.) was used for color development, hematoxylin (Beijing solebao Technology Co., Ltd.) was used for re dyeing, and the slices were washed with distilled water, and conventional dehydration, transparency and sealing were performed. Five visual fields were randomly selected under the microscope, and the proportion of positive cells (brown) was counted under ×200 times.
Cell culture and grouping
The hepatic cancer cell line HepG2 was purchased from ATCC (USA). HepG2 cells were cultured in DMEM medium (Gibco, USA) in an incubator (37°C, 5% CO2) at 100% humidity. Cells were washed in PBS every day, the medium was changed to fresh medium, and the cells were passaged at 80–90% confluence. The HepG2 cells were divided into four groups: NC group, in which cells were not treated; BL group, in which cells were transfected with empty vector; siLINC00152, in which cells were transfected with siLINC00152; and siLINC00152 + EGFR agonist, in which cells were transfected with siLINC00152 and EGFR agonist (Gibco, USA).
Cell proliferation test
Cell proliferation in different groups was measured using CCK-8. HepG2 cells were plated evenly in 96-well plates (3000 per well). Twenty-four hours later, Lipofectamine 2000 was added in accordance with the instructions of transfection to each set of four wells, before and after transfection, respectively (0 h and 48 h), in the corresponding well with 10 µl CCK-8 reagent. Automatic ELISA was used for measurement at 450 nm wavelength. The light absorption value of the well (A) was taken to reflect the cell growth activity. The experiment was repeated three times.
Cell apoptosis by flow cytometry
The apoptosis rate of the transfected cells was detected by flow cytometry. The HepG2 cells were inoculated with 2 × 105 cells per well in six wells, and the cells and culture medium were collected 48 h after transfection. Cells were washed with PBS, 500 µl binding buffer was added to the suspension cell, and then the suspension was mixed into 5 l AnnexinV-FITC and 10 l PI. Room temperature was avoided. After 10 min, the rate of apoptosis was detected by flow cytometry, expressed as a percentage. The experiment was repeated three times.
Cell cycle by flow cytometry
1 × 106 HepG2 cells in different groups were collected, the cells fixed with 70% EtOH overnight, stained with PI for 30 min, and the distribution of the cell cycle was detected by flow cytometry. The process was repeated three times, and the results were expressed as the mean.
Cell invasion by Transwell assay
Hepatic cancer cell line HepG2 cells were digested into a single-cell suspension, washed in serum-free medium three times, and the cell suspension concentration adjusted to 2 × 105/ml reserve. Matrigel was pre-diluted with serum-free DMEM medium at a ratio of 1 : 3, uniformly covered with polycarbonate film on the Transwell chamber, and placed at room temperature for 1 h to solidify the Matrigel into a matrix membrane. The Transwell connected the chamber of 700 l DMEM with 10% foetal bovine serum culture medium. A small indoor inoculation of 200 l serum-free cell-suspension culture was created, the Transwell film was removed after 18 h, and cells were washed twice in pre-cooled PBS, fixed in 4% paraformaldehyde for 15 min, stained in 0.1% crystal violet for 5–10 min, and observed with a microscope through the statistics of Transwell film camera. The cell number was counted.
Cell-migration measurement using a wound-healing assay
The HepG2 cells were inoculated with logarithmic cells, and the cells were inoculated in a six-well plate at the appropriate density. DMEM medium containing 2% serum was incubated in an incubator at 37°C. When the cultured cells grew to a confluence of 80%, a wound was made on the surface of the cell with 200 l gun heads. After 0 and 48 h, respectively, the cells were observed with an inverted microscope, and the distance between the wounds was recorded.
Relative protein expression measurement by WB assay
The different groups of cells treated with different methods were collected. Cell lysates were added to extract the total protein, and the protein concentration was measured using BCA methods following the kit instructions. Next, SDS-PAGE protein electrophoresis was performed. After electrophoresis, the gel was carefully removed and transferred to PVDF film. Five percent skimmed milk powder was sealed for 1 h, placed in EGFR, PI3K, AKT, P21, MMP-2/9, and GAPDH antibody solutions at a dilution of 1 : 1000, and incubated overnight at 4°C. The following day, membranes were washed in TBST three times and placed in secondary antibody solution to incubate for 1 h at room temperature before staining and scanning.
Tumour bearing test
Model preparation and grouping
Normal saline was used to regulate HepG2 cells to 1 × 107/ml, and a HepG2 tumour-bearing model of mice was established by subcutaneous inoculation with 0.2 ml in the right mouse armpit. After 24 h, the nude mice were divided into four groups, with nine nude mice in each group as follows: normal control (NC) group – the nude mice were injected with empty vector in the caudal vein; BL group – the nude mice were injected with siLINC00152 in the caudal vein; siLINC00152 group – the nude mice were injected with siLINC00152 in the caudal vein; and siLINC00152 + EGFR agonist group – the nude mice were injected with siLINC00152 and EGFR agonist in the caudal vein. After 10 days of continuous injection the injections were stopped and the nude mice were killed by dislocation of the spine.
TUNEL assay
According to the literature [8] method, paraffin sections of tumour tissues and cell-apoptosis were detected with TUNEL staining performed according to the instructions of the kit. Under the microscope, images were collected, and Image-Pro Plus 6 image analysis software was used to calculate the total number of cells, the number of apoptotic cells in eight visual fields, and the apoptotic rate.
Statistical analysis
All statistical analyses were performed using SPSS 20.0 (SPSS, USA). The expression level of LINC00152 in tumour specimens was compared with adjacent non-tumour specimens using a paired-samples t-test. The expression differences between cell-cycle, apoptosis, migration, and invasion assays were analysed using an independent samples t-test. All data were presented as mean ± standard deviation. All p-values were two-sided, and p-values of less than 0.05 were deemed statistically significant.
Results
Clinical data
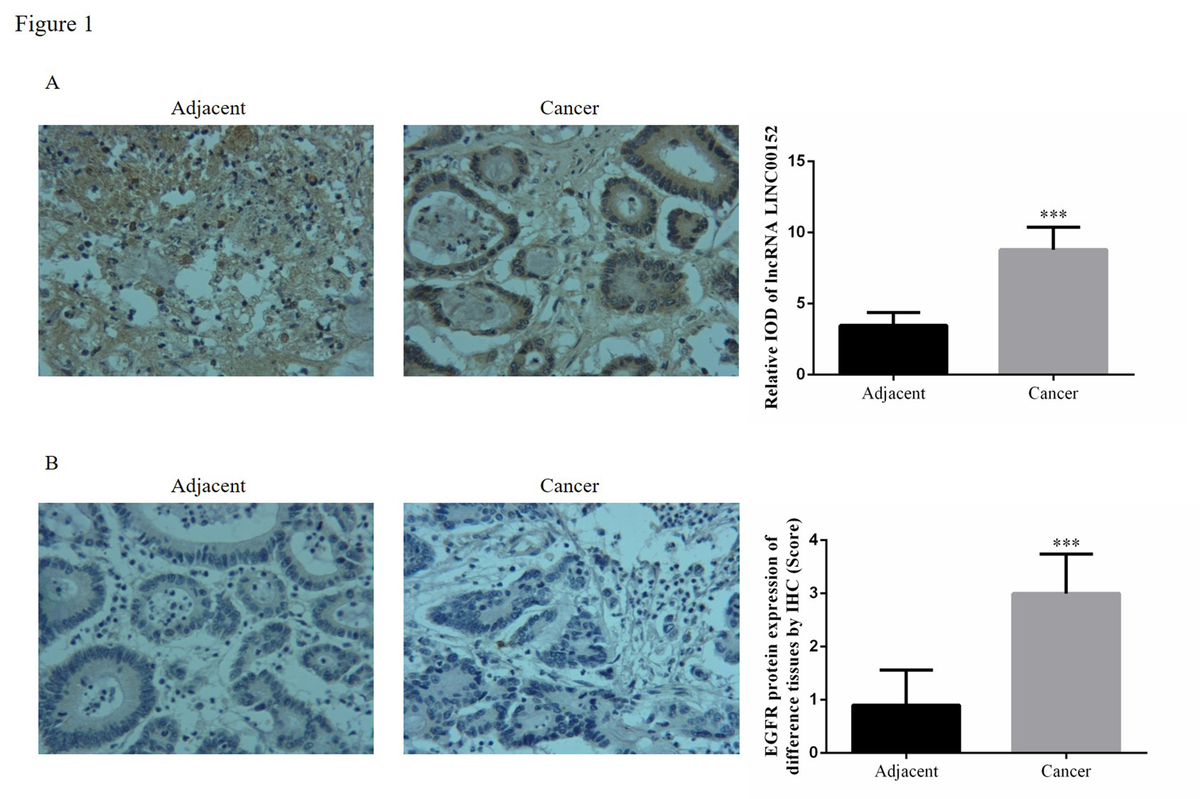
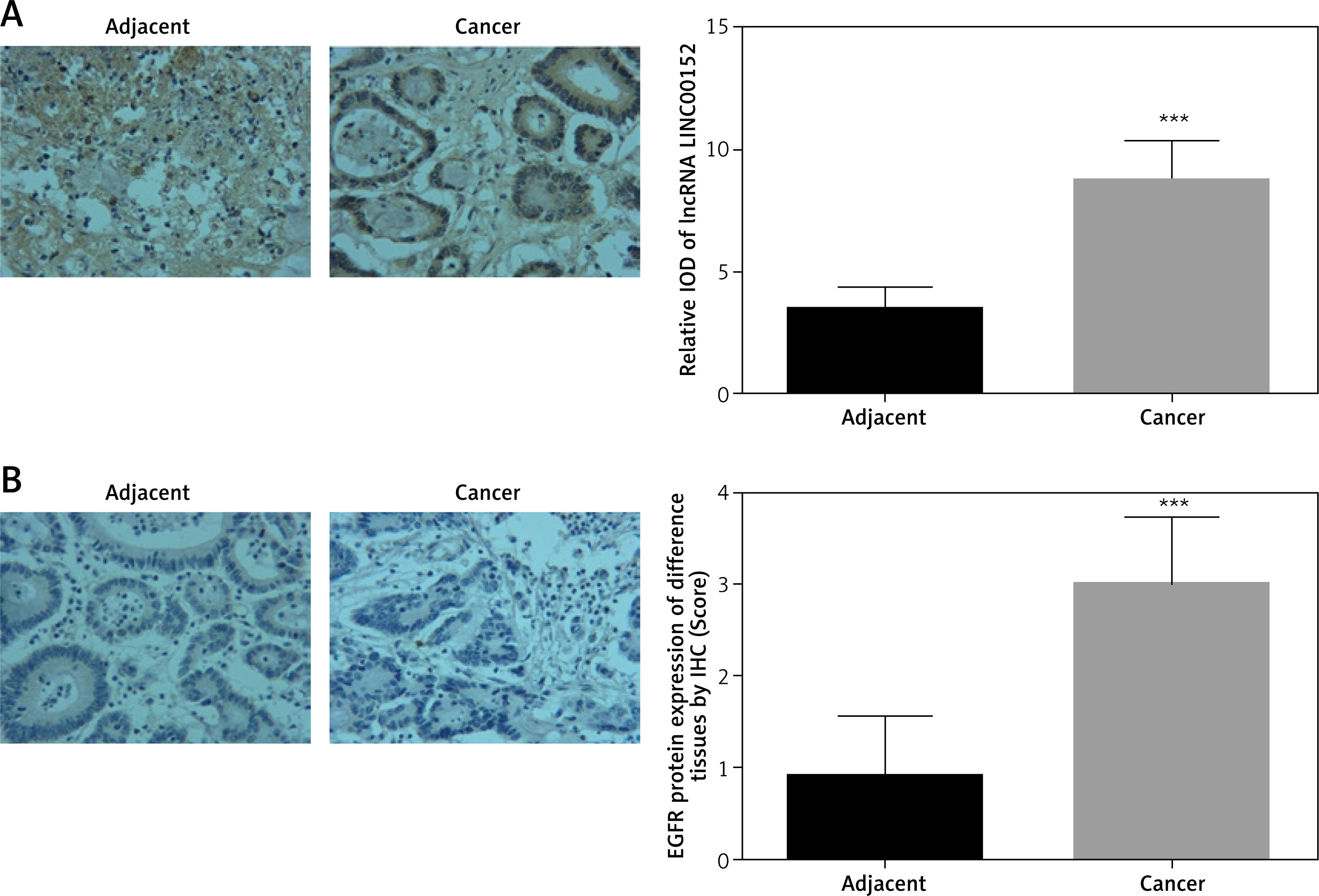
According to the ISH assay, the lincRNA, LINC00152 expression in cancer tissues was significantly increased compared with adjacent normal tissues (p < 0.05). Compared with adjacent normal tissues, EGFR protein expression in cancer tissues was up-regulated according to the IHC assay (p < 0.05). The relative data are shown in Figure 1.
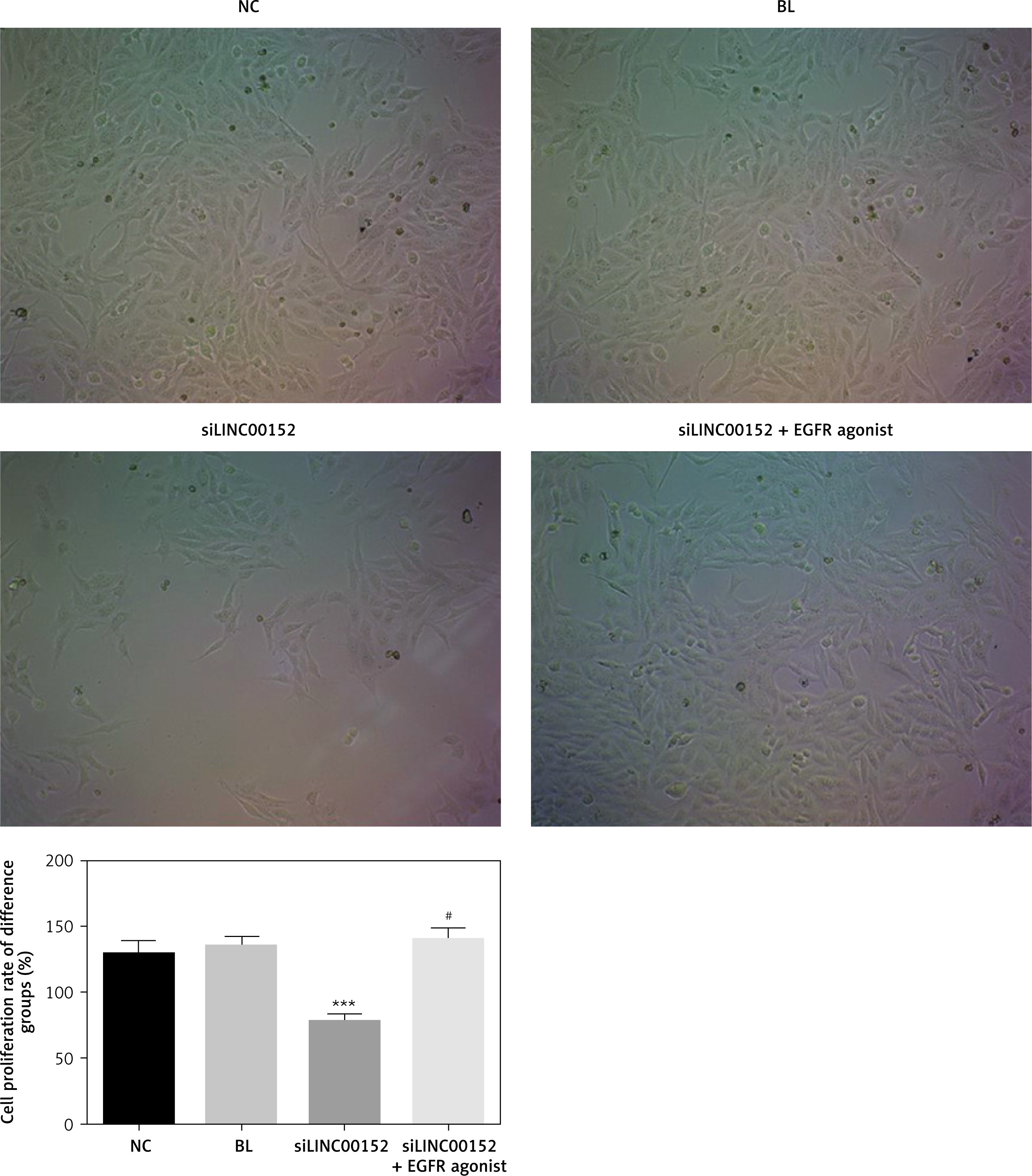
lncRNA LINC 00152 affects HepG2 cell growth in vitro
Compared with the NC group, the cell proliferation rate of the siLINC00152 group was significantly down-regulated (p < 0.05); however, the cell-proliferation rate of the siLINC00152 + EGFR agonist group was significantly up-regulated compared with that of siLINC00152 (p < 0.05), and no significant differences were found compared with the NC group (p > 0.05). The corresponding data are shown in Figure 2.
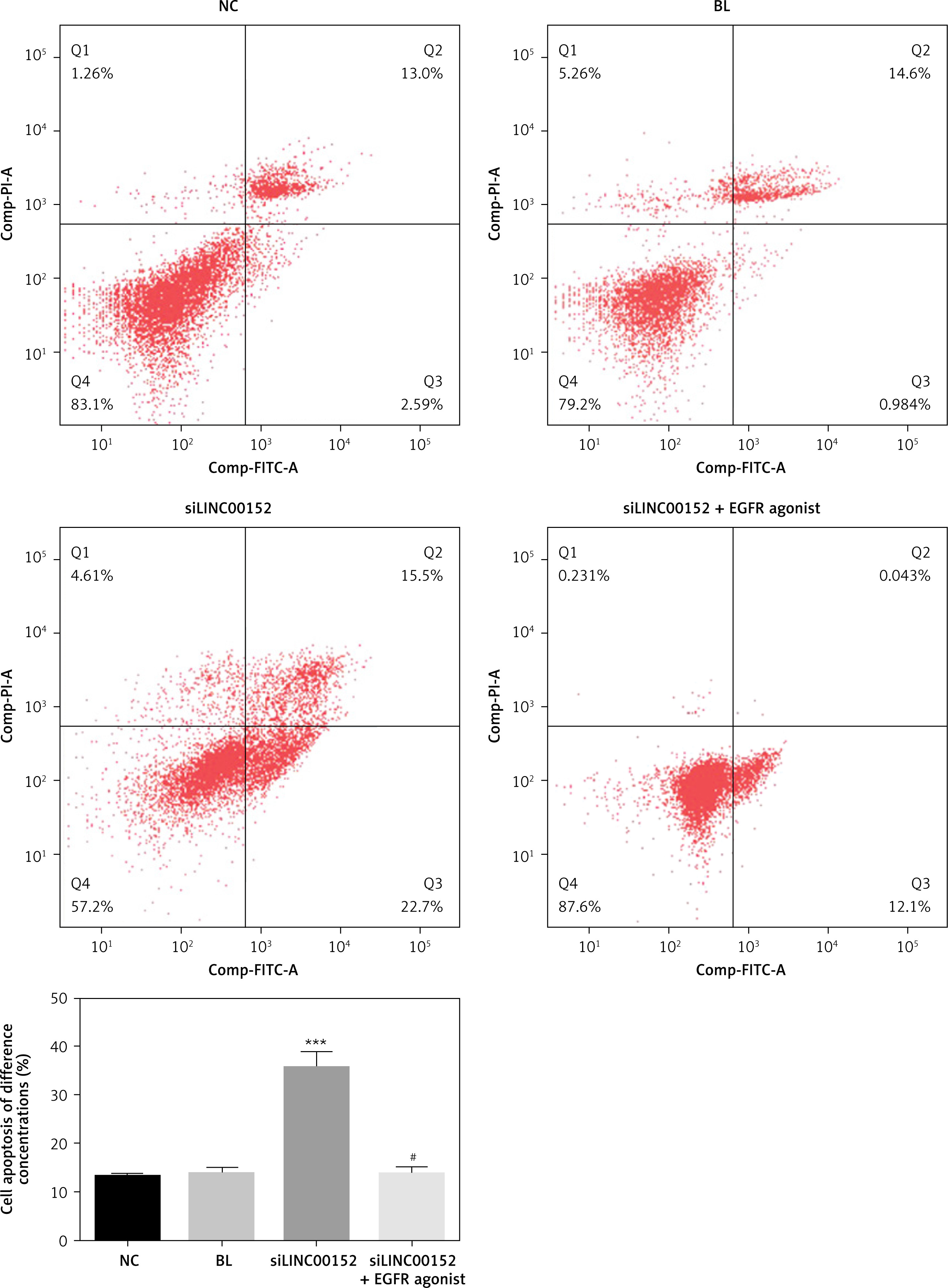
lncRNA LINC 00152 affects HepG2 cell apoptosis in vitro
Compared with the NC group, the cell apoptosis rate in the siLINC00152 group was significantly up-regulated (p < 0.05); however, the cell apoptosis rate in the siLINC00152 + EGFR agonist group was significantly down-regulated compared with that of siLINC00152 (p < 0.05), and no significant differences were found compared with the NC group (p > 0.05). The corresponding data are shown in Figure 3.
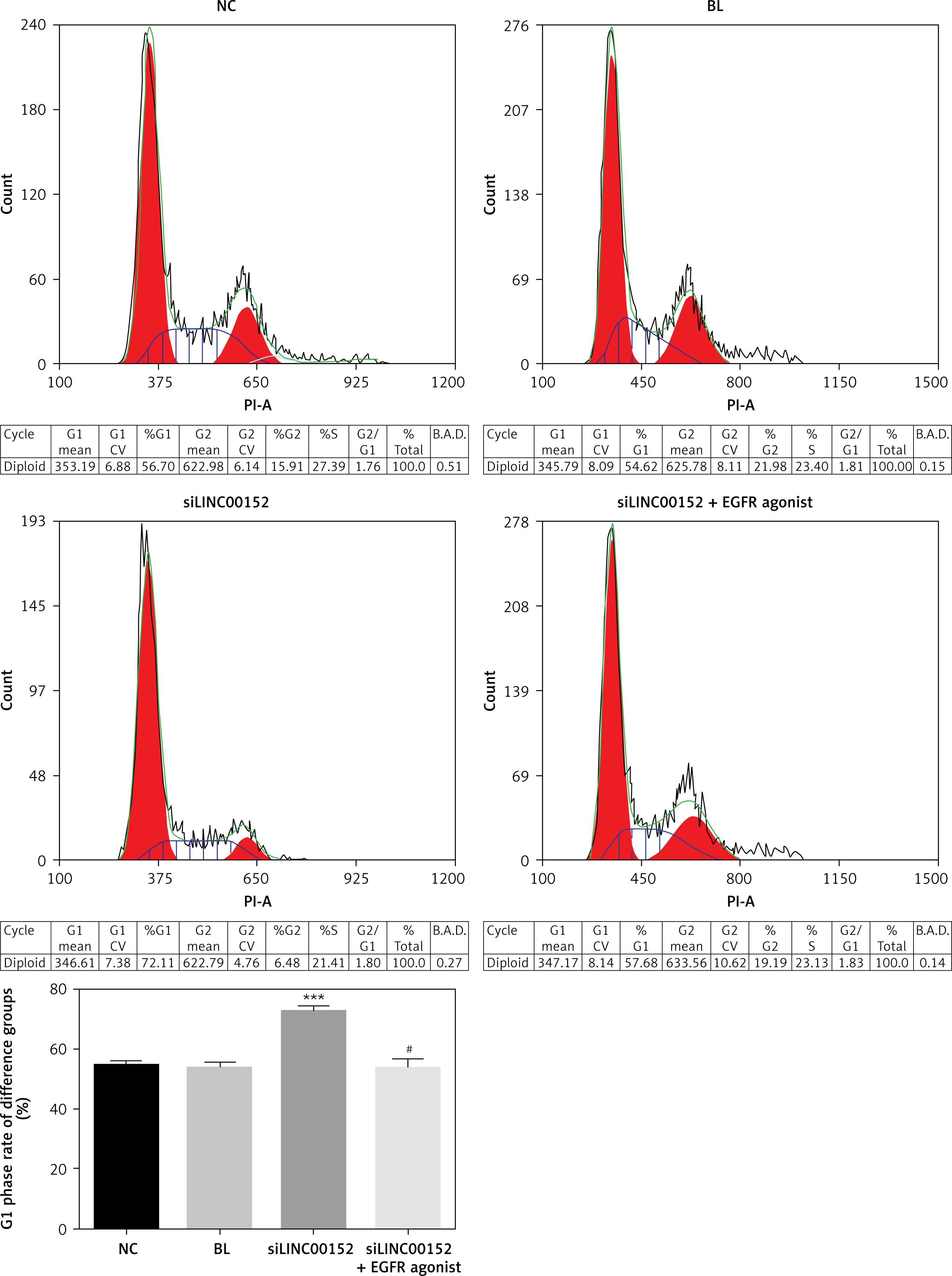
lncRNA LINC 00152 affects HepG2 cell cycle in vitro
Compared with the NC group, the G1 phase rate in the siLINC00152 group was significantly up-regulated (p < 0.05); however, the G1 phase rate of the siLINC00152 + EGFR agonist group was significantly down-regulated compared with that of siLINC00152 (p < 0.05), and no significant differences were found compared with the NC group (p > 0.05). The corresponding data are shown in Figure 4.
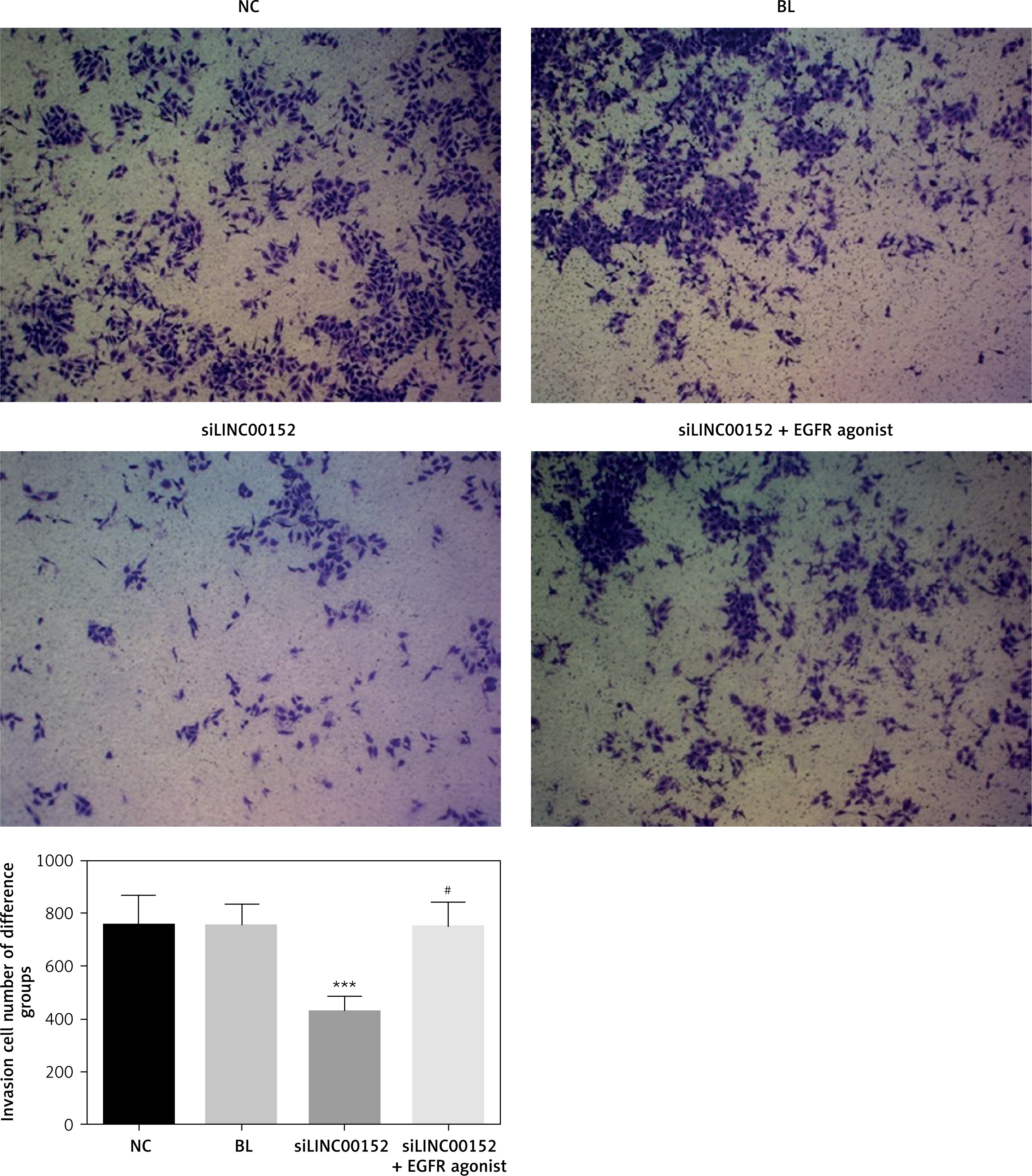
lncRNA LINC00152 affects HepG2 cell invasion in vitro
Compared with the NC group, the invasion HepG2 cell number in the siLINC00152 group was significantly down-regulated (p < 0.05); however, the invasion HepG2 cell number in the siLINC00152 + EGFR agonist group was significantly up-regulated compared with that of siLINC00152 (p < 0.05), and no significant differences were found compared with the NC group (p > 0.05). The corresponding data are shown in Figure 5.
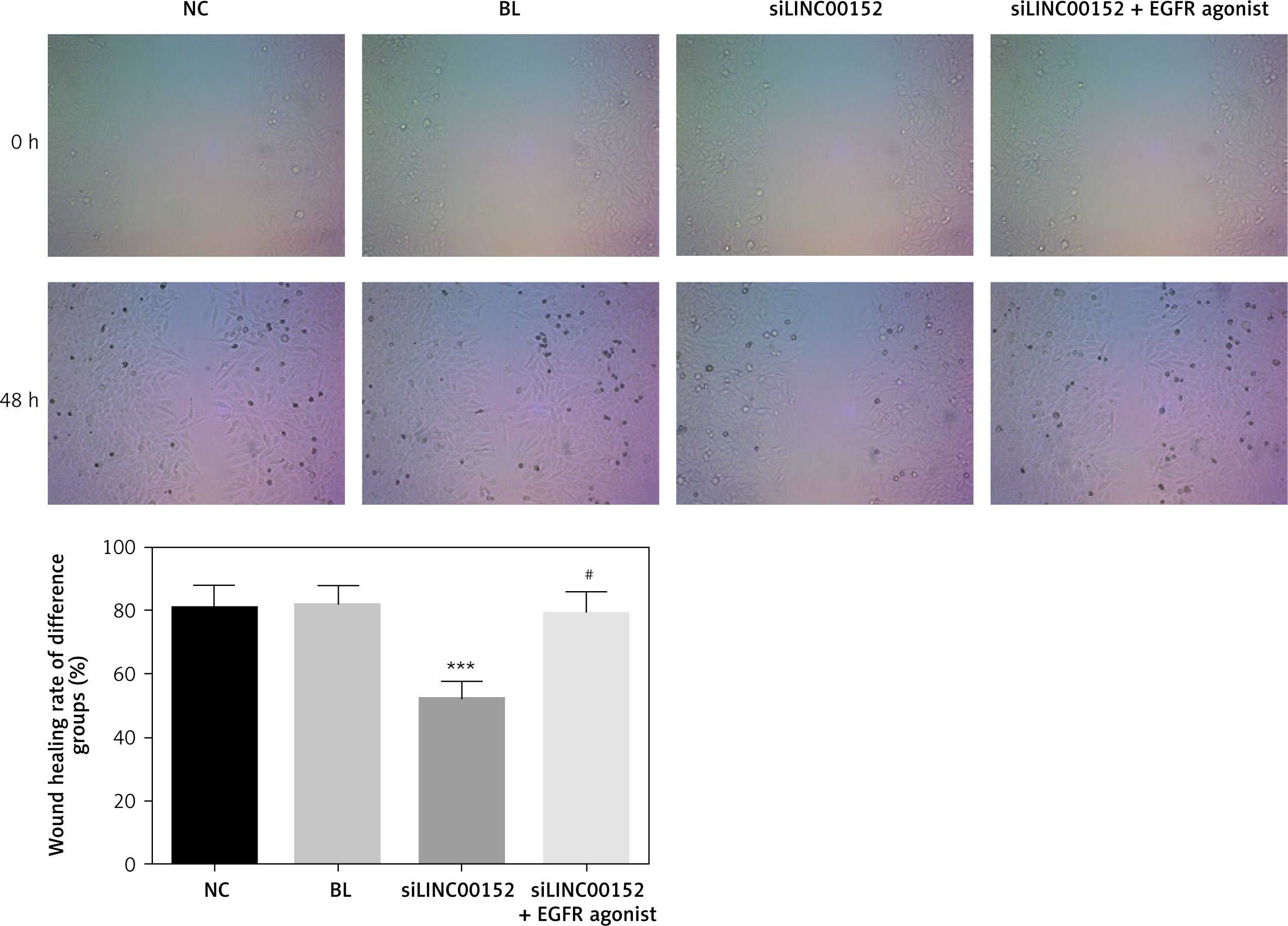
lncRNA LINC00152 affects HepG2 cell migration in vitro
Compared with the NC group, the wound-healing rate in the siLINC00152 group was significantly down-regulated (p < 0.05); however, the wound-healing rate in the siLINC00152 + EGFR agonist group was significantly up-regulated compared with that of siLINC00152 (p < 0.05), and no significant differences were found compared with the NC group (p > 0.05). The corresponding data are shown in Figure 6.
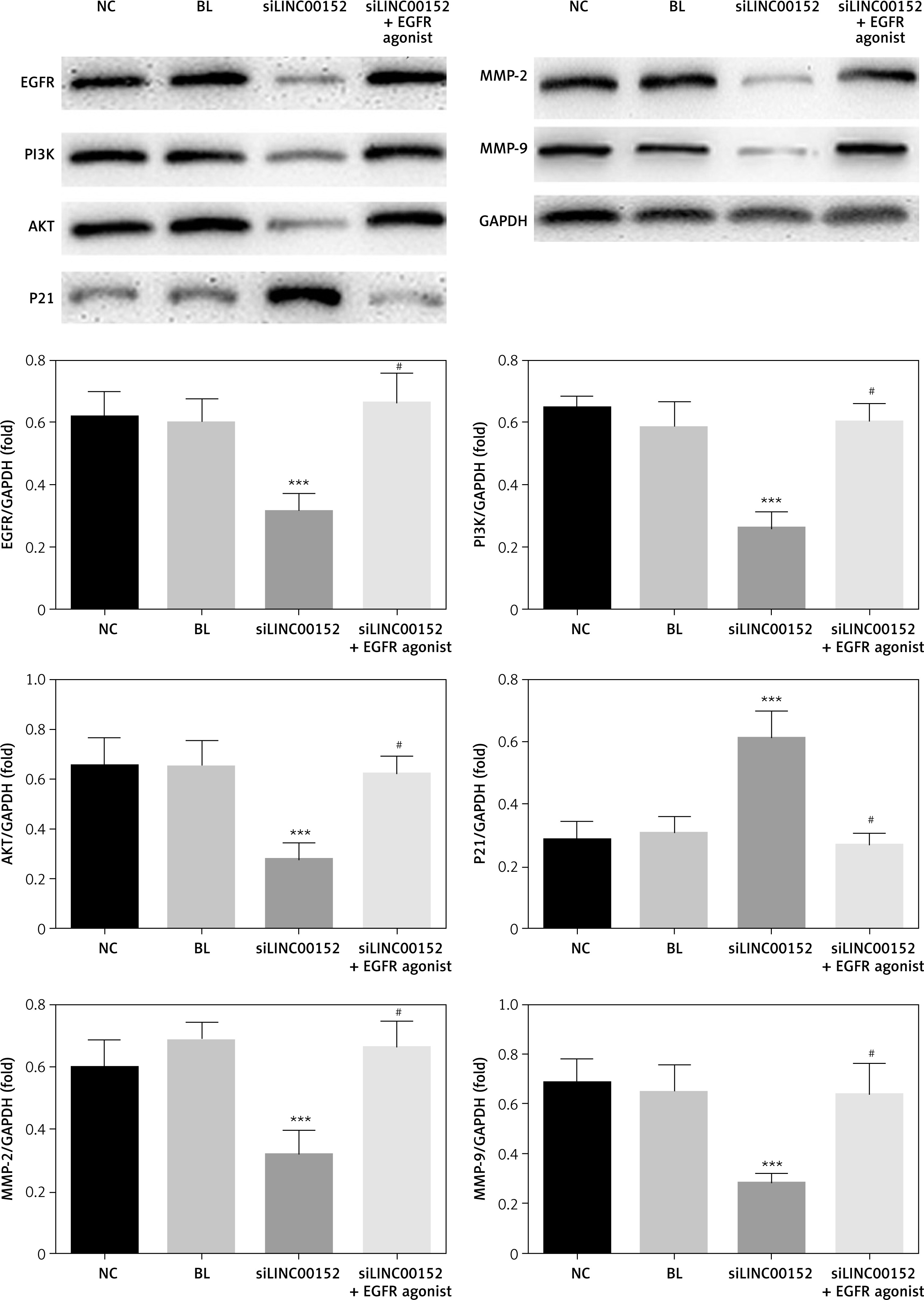
The relative expression of proteins according to WB assay
Compared with the NC group, EGFR, PI3K, AKT, MMP-2, and MMP-9 protein expression in the siLINC00152 group was significantly reduced, and P21 protein expression in the siLINC00152 group was significantly increased (p < 0.05). However, EGFR, PI3K, AKT, MMP-2, and MMP-9 protein expression in the siLINC00152 + EGFR agonist group was significantly up-regulated, and P21 protein expression in the siLINC00152 + EGFR agonist group was significantly down-regulated, compared with the siLINC00152 group (p < 0.05 in both cases). The corresponding data are shown in Figure 7.
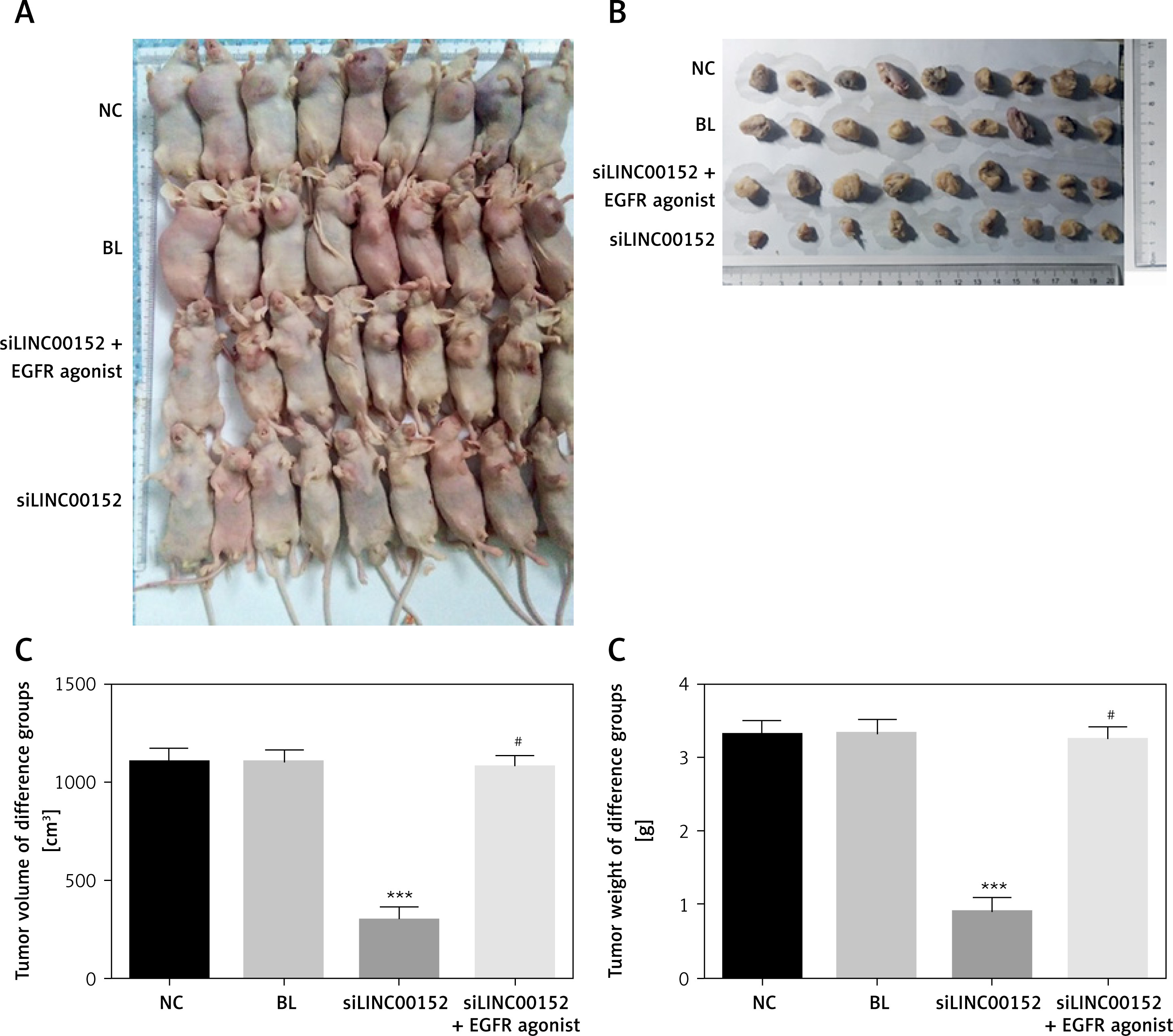
Tumour volume and weight in different groups
The tumour volume and weight of the siLINC00152 group were significantly suppressed compared with those of the NC group (p < 0.05 in both cases); compared with the siLINC00152 group, the tumour volume and weight of the siLINC00152 + EGFR agonist group were significantly up-regulated (p < 0.05 in both). The corresponding data are shown in Figure 8.
Figure 8
The tumour of different groups. A – The tumours of different groups in vivo. B – Taking out the tumour tissues of different groups. C – The tumour volume of different groups. D – The tumour weight of different groups
***P < 0.05, compared with the NC group, #p < 0.05, compared with the siLINC00152 group.
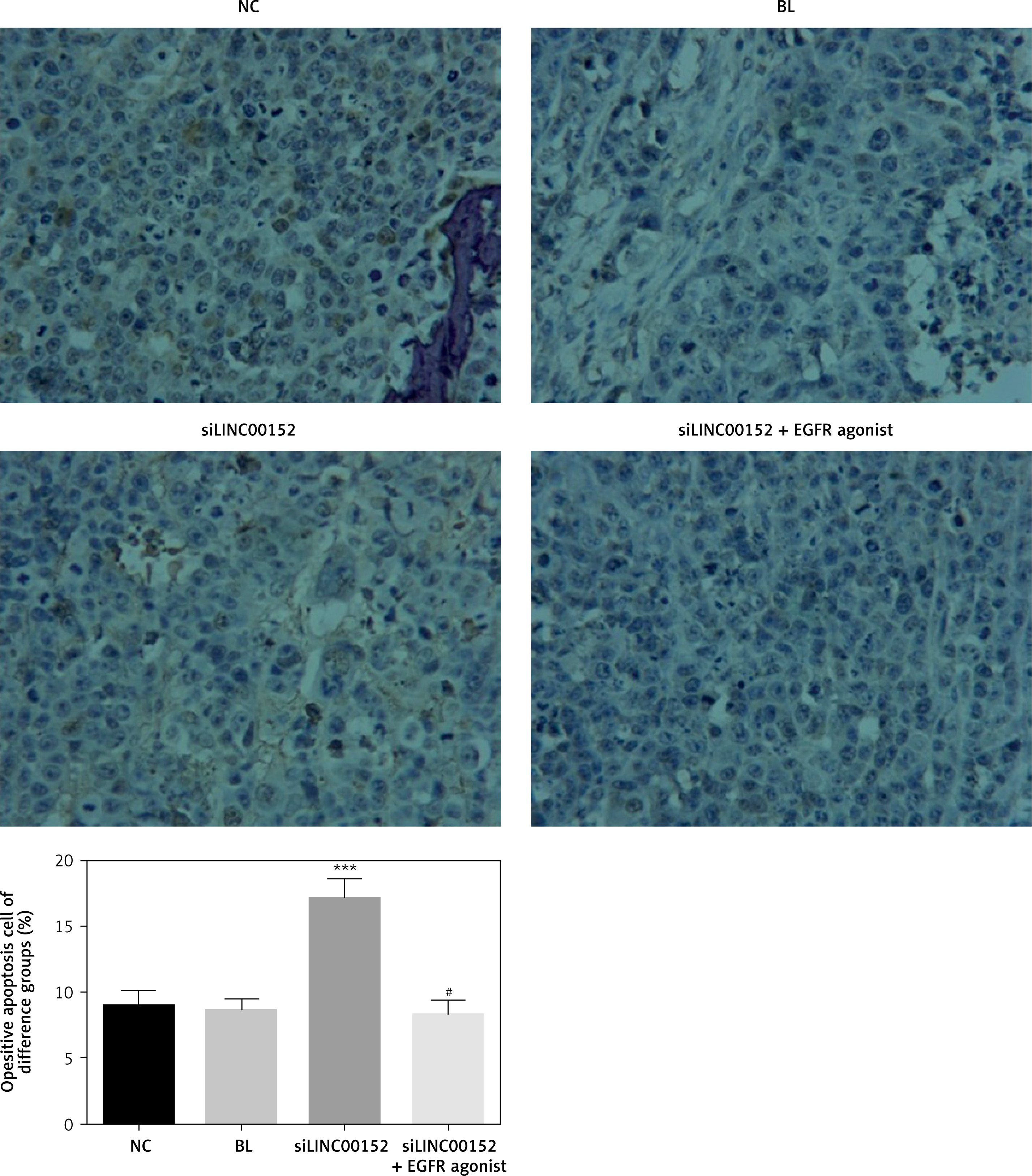
Cell apoptosis in different groups in vivo
Compared with the NC group, the cell apoptosis rate of the siLINC00152 group was significantly up-regulated according to the TUNEL assay (p < 0.05). However, the cell apoptosis rate in the siLINC00152 + EGFR agonist group was significantly down-regulated compared with the siLINC00152 group according to the TUNEL assay (p < 0.05). The corresponding data are shown in Figure 9.
The relative expression of proteins by IHC in vivo
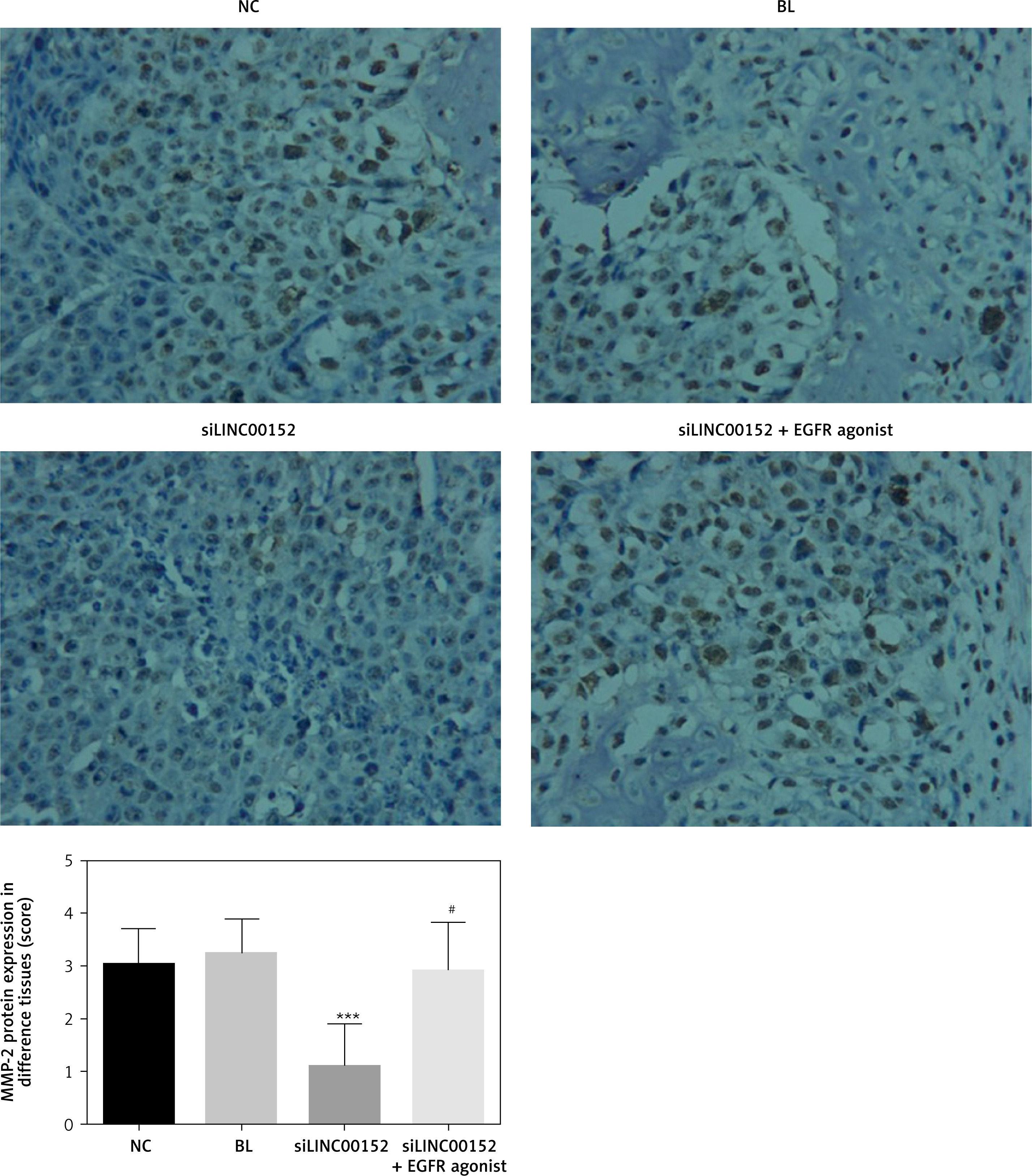
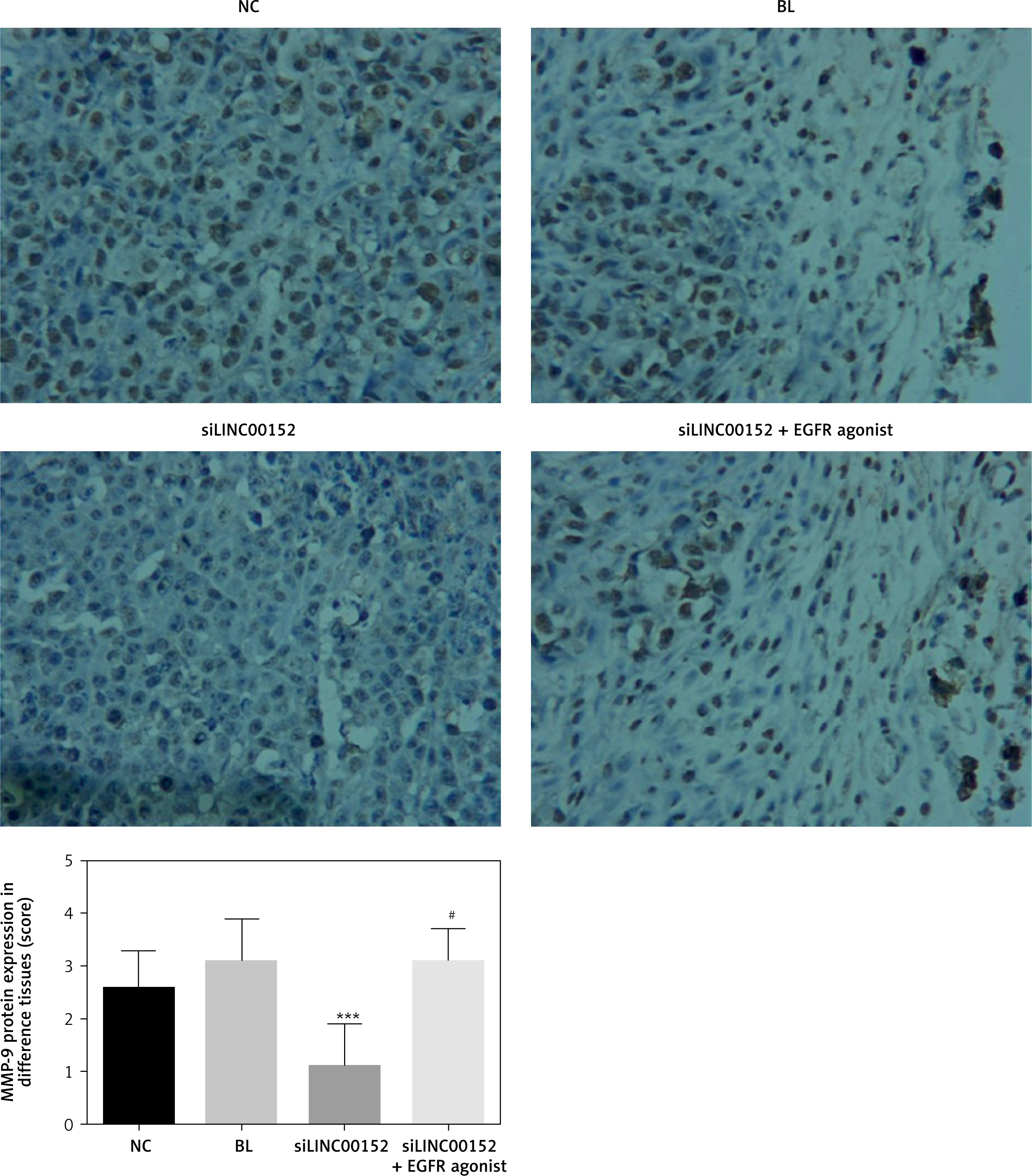
Compared with the NC group, EGFR, PI3K, AKT, MMP-2, and MMP-9 protein expression in the siLINC00152 group was significantly reduced, and P21 protein expression in the siLINC00152 group was significantly increased (p < 0.05 in both cases). However, EGFR, PI3K, AKT, MMP-2, and MMP-9 protein expression in the siLINC00152 + EGFR agonist group was significantly up-regulated, and P21 protein expression in the siLINC00152 + EGFR agonist group was significantly down-regulated compared with the siLINC00152 group (p < 0.05 in both cases). The corresponding data are shown in Figures 10–15.
Figure 10
The EGFR protein expression of different groups by IHC
***P < 0.05, compared with the NC group, #p < 0.05, compared with the siLINC00152 group.
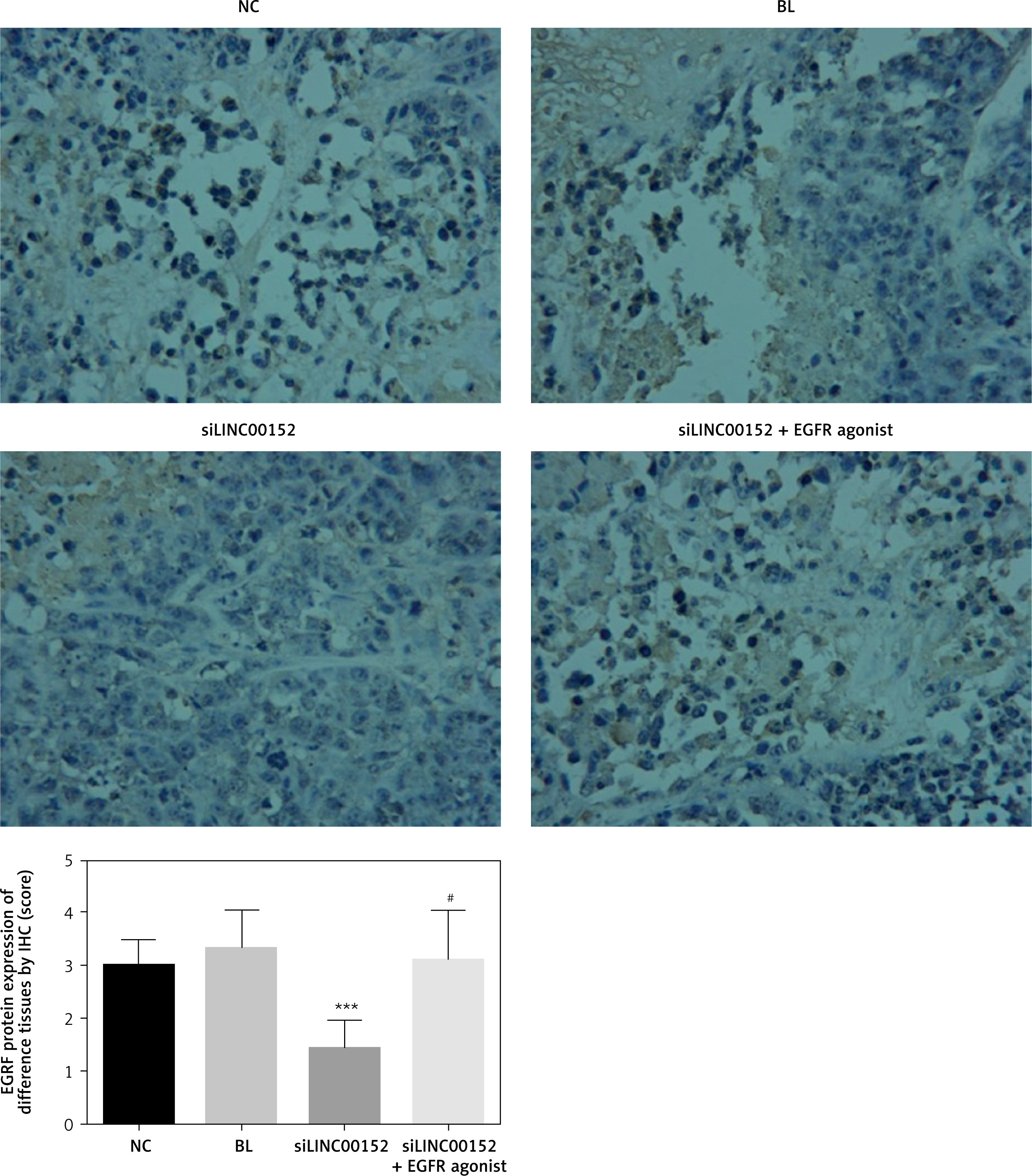
Figure 11
The PI3K protein expression of different groups by IHC
***P < 0.05, compared with the NC group, #p < 0.05, compared with the siLINC00152 group.
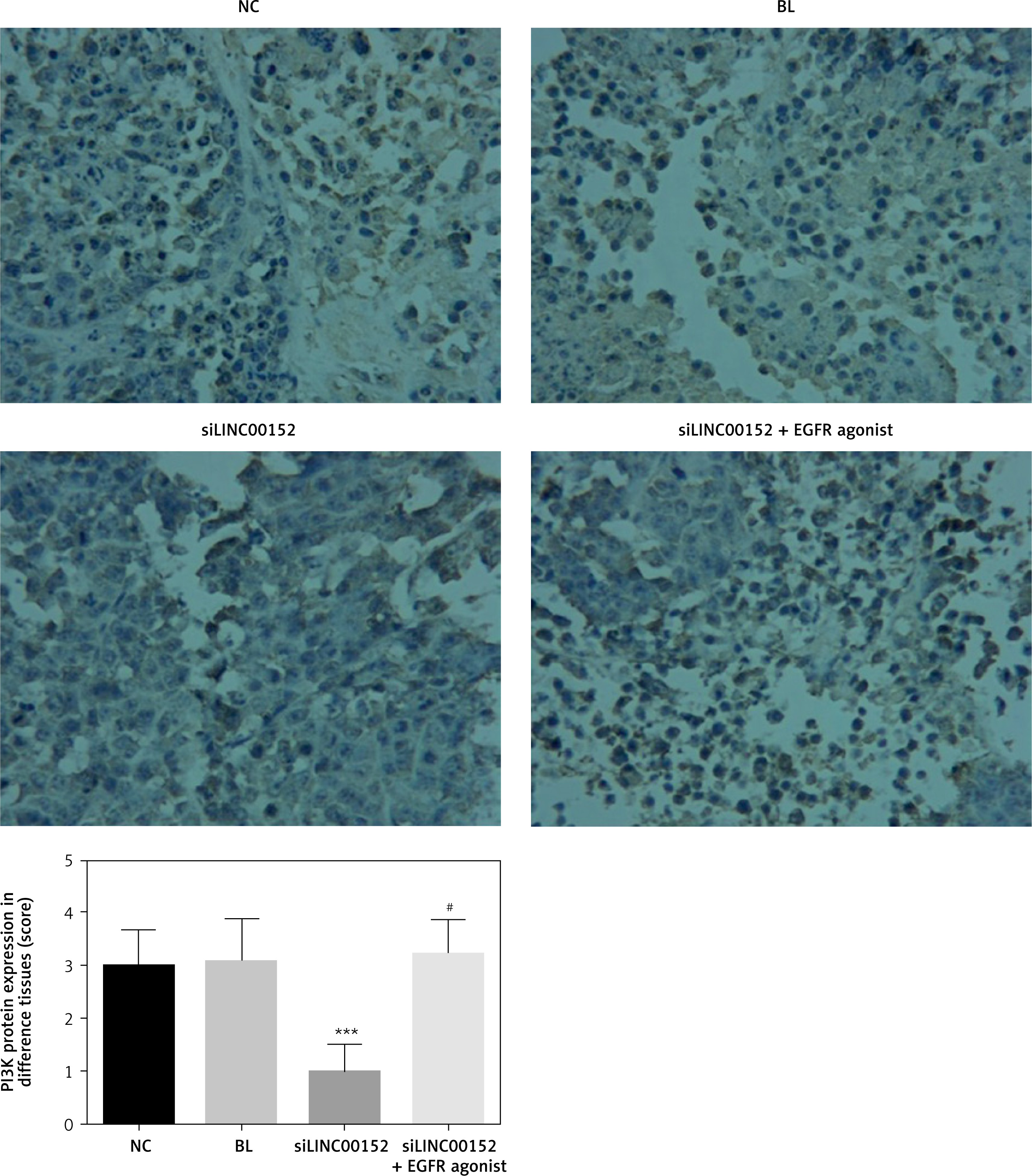
Figure 12
The AKT protein expression of different groups by IHC
***P < 0.05, compared with the NC group, #p < 0.05, compared with the siLINC00152 group.
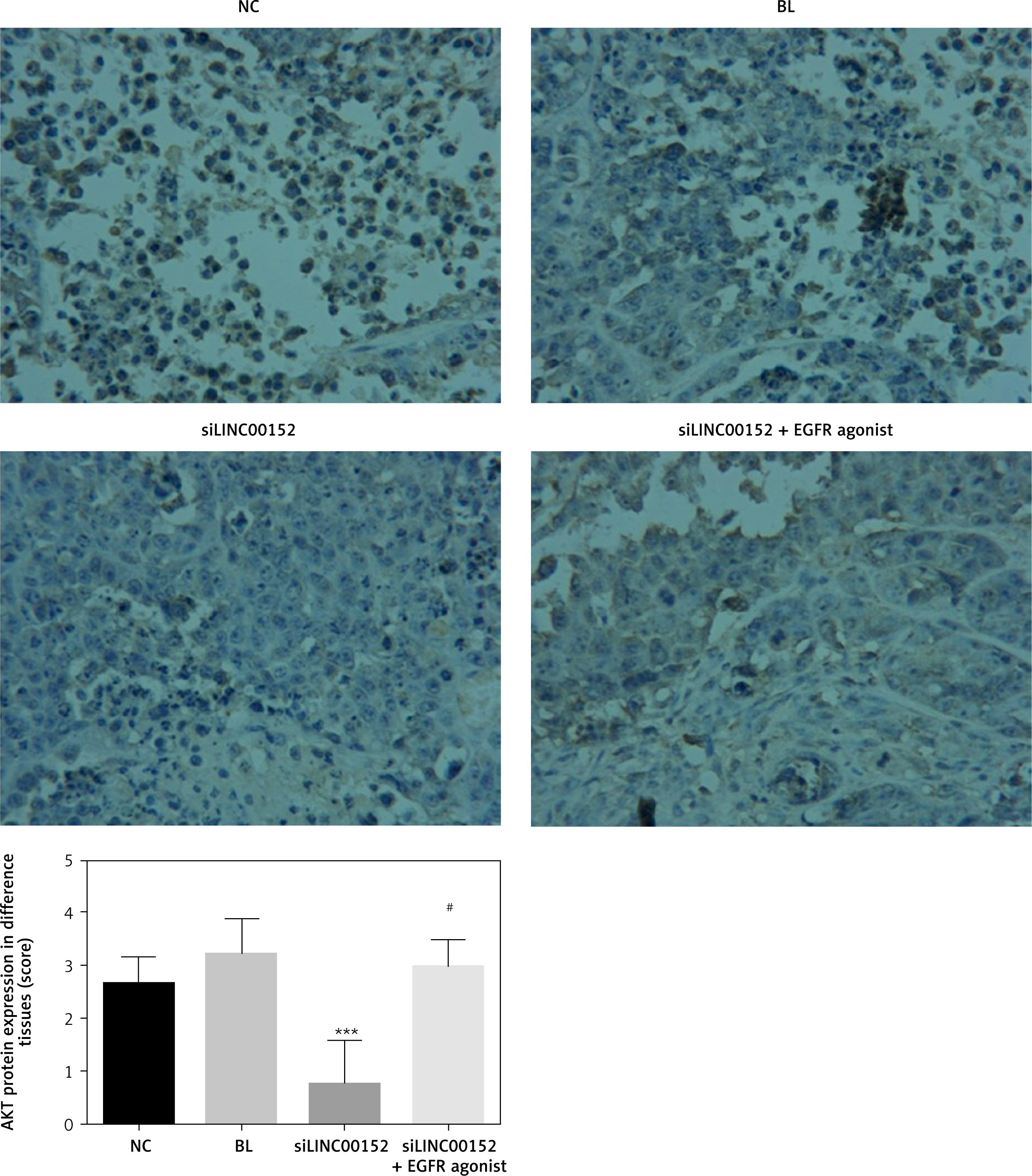
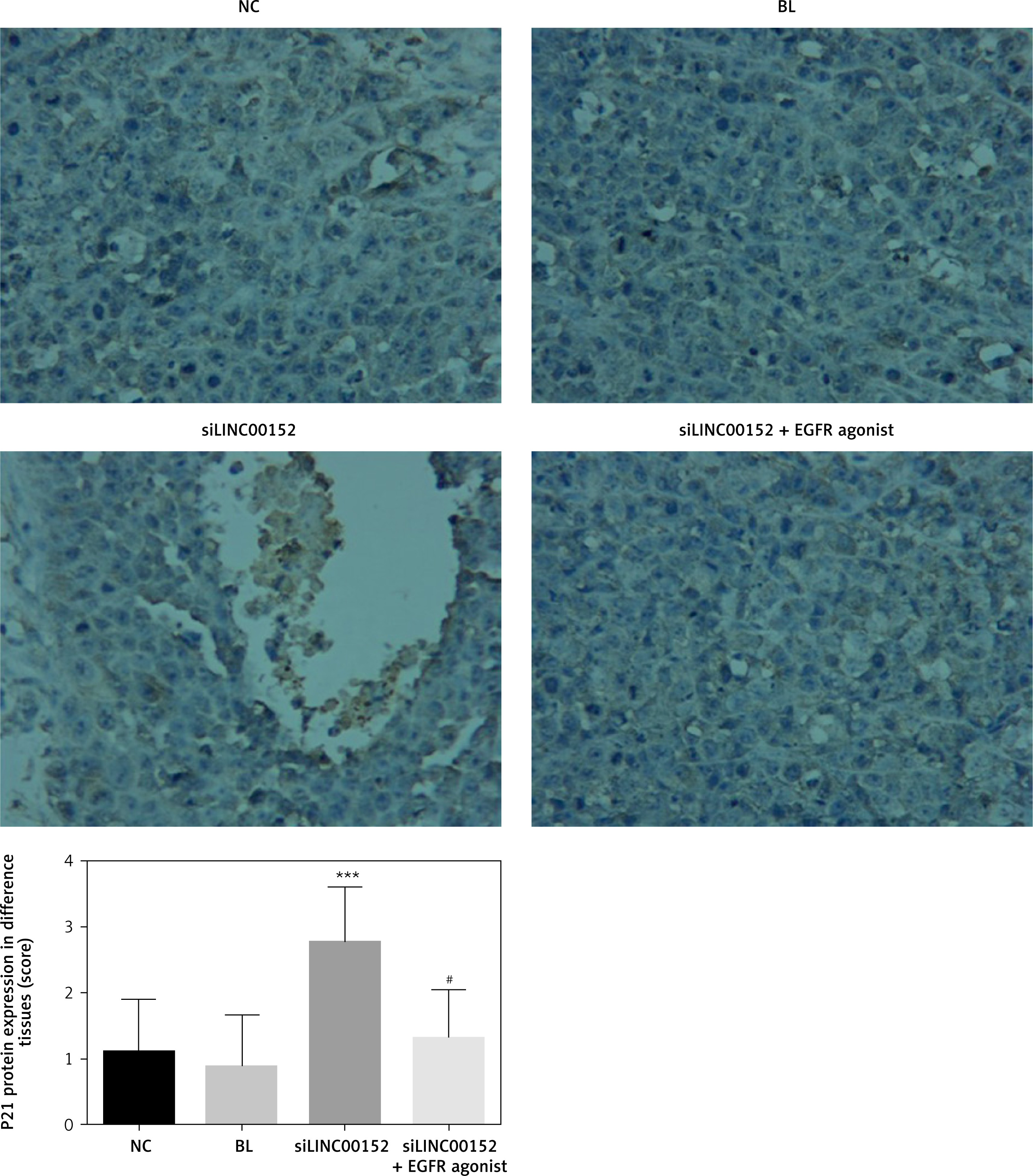
Figure 13
The P21 protein expression of different groups by IHC
***P < 0.05, compared with the NC group, #p < 0.05, compared with the siLINC00152 group.
Discussion
With the rapid development of gene-chip and high-throughput sequencing technologies, the discovery of lncRNA has enabled a new phase in the study of the human genome. Previous studies have found that some lncRNAs are closely correlated with disease [9, 10]. In situ hybridisation, as an intuitive test of target DNA or RNA in tissue cells, has high sensitivity and specificity and can be used as a semi-quantitative detection method. LINC00152 belongs to the class of intergenic lncRNA, whose gene is located in ch2p11.2 and has three transcriptional variants, with lengths of 518 nt, 664 nt, and 828 nt. In our present study, we found that lncRNA LINC 00152 expression in hepatic cancer tissues was significantly up-regulated compared with that of adjacent normal tissues. Previous studies have found that EGFR is closely correlated with LINC 00152 in some cancers [11–13]. However, the correlation between LINC00152 and EGFR and the effects of LINC00152 in hepatic cancer remain unclear. In our clinical data, we found that LINC 00152 expression and EGFR protein expression were significantly up-regulated compared with adjacent normal tissues. Next, we wanted to explain the effects and mechanism of LINC00152 in development and the progression of hepatic cancer.
Previous studies have reported that lncRNA LINC00152 is highly expressed in solid tumours, such as gastric cancer, renal cell carcinoma, gallbladder cancer, and glioma [14–16]. In our present studies, we knocked-down the lncRNA LINC00152 by si-LINC00152 to evaluate biological cancer activities. The results showed that lncRNA LINC00152 knockdown had the effect of suppressing cell proliferation, cell invasion and migration, and improving cell apoptosis by maintaining the cell cycle in the G1 phase in the in vitro assay and inhibiting tumour growth in the vivo assay. In the following study, we wanted to explain the mechanism of siLINC00152, for which we knocked-down lncRNA LINC00152 at the molecular level.
EGFR is one of the members of the tyrosine-receptor family, which consists of three parts: the extracellular ligand-binding region, a transmembrane region, and an intracellular region. The receptor tyrosine kinase (RTK) system of the intracellular region is key to its function [17]. EGFR is not activated in its monomeric form and, when the ligand is bound, can form homo or heterodimers, causing activation of RTK receptor autophosphorylation to promote intracellular tyrosine residues, further activation of downstream signalling pathways (including protein kinase (BAKT) and mitogen-activated protein kinase (MAPK) signal transduction), and regulation of cell biological behaviour [18, 19]. A great deal of evidence suggests that E, G, F, and R play important roles in the occurrence and development of malignant tumours [20]. Breast, ovarian, colon, bladder, and other tumours show that the abnormal expression of EGFR tumour accounts for a large proportion of cancers. High expression of EGFR can cause cell proliferation, invasion, and metastasis; promote tumour angiogenesis; and inhibit tumour-cell apoptosis [21]. The occurrence and development of some malignant tumours in humans are related to the abnormality of the PKB/AKT signalling pathway. Studies have shown that the EGFR/AKT signalling pathway is involved in regulating the apoptosis, proliferation, differentiation, and metabolism of cells [22–24]. In our present study, we find that EGFR and its downstream genes, PI3K and AKT, were significantly suppressed compared to LINC00152 knock-down by siLINC00152. We inferred that those might the results that the mechanism of siLINC00152 to regulate hepatic cancer biological activities (cell proliferation, apoptosis, cell cycle, invasion and migration) in in vitro and in vivo studies.
P21 is a broad-spectrum cyclin an inhibitor of cyclin kinase (CDK). It regulates cell-cycle progression and participates in cell growth, differentiation, ageing, and death. Most studies have shown that p21WAF1/CIP1 plays an important role in the process of cell senescence. The expression of p21 in transformed cells plays a key role in growth arrest [25]. Up-regulation of p21 levels can inhibit the activity of CDK and inhibit gene expression. The cells cannot enter the S phase and eventually undergo cell-cycle arrest [26]. In our present study, we infer that siLINC00152 stimulates P21 expression by suppressing the EGFR/PI3K/AKT signalling pathway to maintain the cell cycle in the G1 phase.
Matrix metalloproteinases (MMPs) comprise a large family of enzymes that can degrade almost any kind of protein components in the extracellular matrix, destroy the histological barrier of tumour cell invasion, and play a key role in tumour invasion and metastasis [27]. Among the many MMPs, MMP-2 and MMP-9 are involved in angiogenesis and are directly related to the degradation of the basal membrane, which is involved in tumour metastasis and prognosis [28, 29]. MMP-2 and MMP-9 are important roles of EGFR/PI3K/AKT downstream genes and closely correlate with tumour invasion and migration [30, 31]. From the results of our present study, we conclude that siLINC00152 suppresses hepatic cancer-cell invasion and migration by suppressing MMP-2 and MMP-9 expression by, in turn, regulating the EGFR/PI3K/AKT pathway.
In conclusion, siLINC00152, which was knocked-down by lncRNA LINC00152, suppresses hepatic cancer-cell biological activities via EGFR/PI3K/AKT, its downstream gene P21, and MMP-2/9 in in vitro and in vivo assays.
In our present study, there were also some limitations; we did not analyse the correlation between lncRNA LINC00152 and clinical pathology and prognosis in hepatic cancer patients.