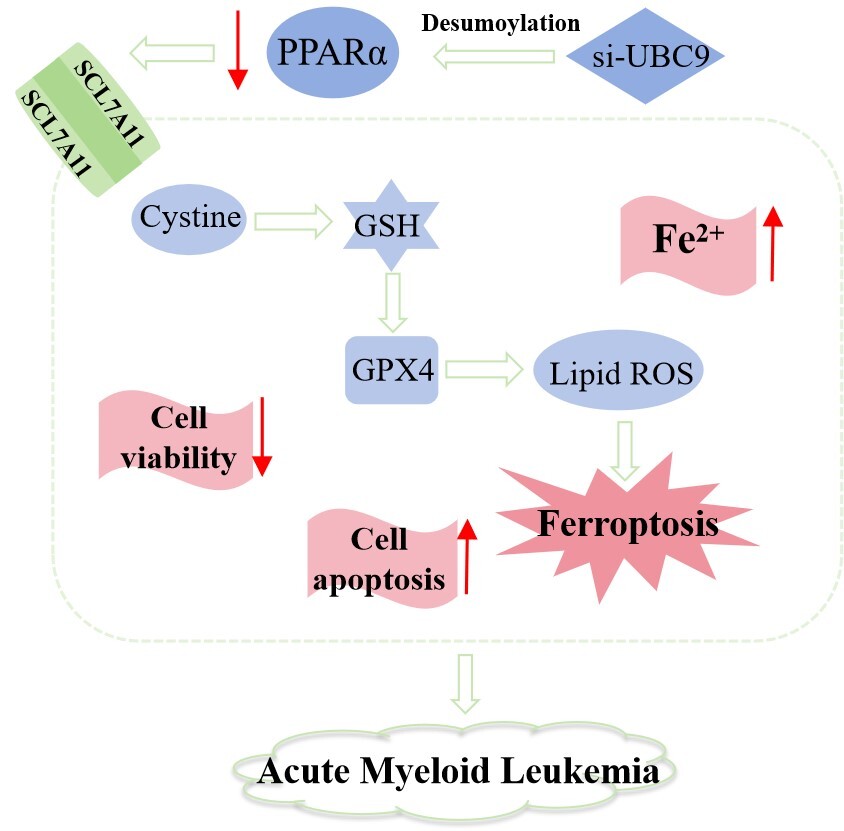
Introduction
Acute myeloid leukemia (AML) develops from the malignant transformation of myeloid lineage stem cell precursors [1], constituting 15% to 20% of leukemias in the United States, with 21,450 cases diagnosed in 2019 [2]. The progression of AML is driven by genetic variations that trigger neoplastic changes and further ensue clonal proliferation [1]. Excessive proliferation and subsequent accumulation of immature myeloid cells in the bone marrow and peripheral blood interfere with the generation of normal blood cells, resulting in abnormal hematopoiesis [3]. AML is a hematologic malignancy characterized by the clonal proliferation of immature myeloid cells in the bone marrow, leading to disruption of normal hematopoiesis and bone marrow failure [4]. Currently, despite advances in therapies for AML, the survival of patients still remains limited, due to disease relapse as a consequence of unhindered cell proliferation following hematopoietic stem cell transplantation [5]. Therefore, novel approaches to effectively suppress expansion of AML cells are urgently needed.
Ferroptosis is a newly identified form of regulated cell death characterized by the accumulation of iron-dependent lipid peroxides to fatal levels [6]. Morphologically, biologically, and genetically separate from apoptosis, ferroptosis is considered to be regulated necrosis [7]. Glutathione peroxidase 4 (GPX4) is a key regulator of ferroptosis and plays a crucial role in converting lipid hydroperoxides to non-toxic lipid [8]. Additionally, inhibition of recombinant solute carrier family 7, member 11 (SLC7A11), the light chain of system xc-, attenuates GSH levels and GPX4 activity, resulting in the accumulation of lethal lipid peroxides and the induction of ferroptosis [9]. Inducing ferroptosis enhances anti-leukemic activity [10] and serves as a mechanism underlying chemotherapy targeting AML [11].
Ferroptosis and cell fate are impacted by genetic cues [12]. Recent improvement in the understanding of the genetic variations in AML progression contributes to development of some promising novel therapies for better outcomes [13]. Adipose triacylglyceride lipase (ATGL) silencing-induced inhibition of AML cell proliferation is concomitant with downregulation of peroxisome proliferator-activated receptor α (PPARα) [14]. PPARα is a ligand-activated transcription factor, belonging to the NR1C nuclear receptor subfamily that has many members implicated in large-scale remodeling of lipid homeostasis [15], which affects the sensitivity of cells to ferroptosis [16]. Additionally, PPARα agonists alleviate erastin-induced ferroptotic death in liver cancer cells [17]. Nevertheless, whether PPARα inhibition blocks AML cell growth through inducing ferroptosis has yet to be verified.
Sumoylation is a reversible post-translational modification modulating protein stability, nuclear-cytosolic transport, and transcriptional regulation [18]. Sumoylation is mediated by small ubiquitin-like modifier proteins (SUMOs), such as SUMO1, which is covalently attached to and detached from other proteins in cells to alter their function [19]. PPARα is susceptible of SUMOylation modification, and deSUMOylating PPARα can promote its ubiquitination-mediated degradation [20]. Through a String-predicted PPARα-protein interaction map, UBE2I (ubiquitin carrier protein 9, UBC9), the only E2 SUMO-conjugating enzyme, was identified as a protein able to interact with PPARα. A previous study documented that knockdown of UBC9 inhibits AML cell proliferation [21].
Accordingly, we hypothesized that blocking SUMOylation modification of PPARα promotes its degradation, which inhibits cell proliferation in AML by facilitating ferroptosis. The present study aimed to test this hypothesis through establishing an in vivo AML model under the influence of a PPARα inhibitor or PPARα agonist plus UBC9 silencing.
Material and methods
Cell culture
Human AML HL-60 cell lines and THP-1 cell lines were purchased from American Type Culture Collection (ATCC; CCL-240 and TIB-202, Manassas, VA, USA). HL-60 cells were cultured in ATCC-formulated Iscove’s Modified Dulbecco’s Medium (30-2005, ATCC, USA), supplemented with 20% fetal bovine serum (FBS; HY-P2352, MedChemExpress, Monmouth Junction, NJ, USA). A complete medium comprising ATCC-formulated RPMI-1640 medium (30-2001, ATCC, USA), 10% FBS, and 0.05 mM 2-mercaptoethanol (21985023, Thermo Fisher, Waltham, MA, USA) was used to maintain THP-1 cells. Cell culture was performed with 5% CO2 at 37°C.
Cell transfection
THP-1 cells were transfected with small interfering RNA targeting UBC9 (siUBC9; SR305005, OriGene, Rockville, MD, USA) or its negative control (siNC; PS100001, OriGene, USA) using Lipofectamine 3000 transfection reagent (L3000015, Thermo Fisher, USA). In short, 1 × 104 THP-1 cells were seeded in each well of 96-well plates and cultured to form an 80% confluent monolayer. The above plasmids and Lipofectamine 3000 transfection reagent were diluted and incubated together for 15 min at 37°C to form gene-lipid complexes. The complexes were then incubated for 48 h with the monolayer, and the transfection efficiency was assessed by quantitative reverse transcription-polymerase chain reaction (qRT-PCR).
QRT-PCR
Total RNA from transfected/nontransfected THP-1 cells was obtained using Trizol reagents (15596026, Thermo Fisher, USA) with a diluted concentration of 100 µg/ml, followed by reverse transcription employing a reverse transcription kit (K1622, Yaanda Biotechnology, Beijing, China) to generate cDNA. QPCR was conducted to achieve cDNA amplification in a PCR System (7500 Fast Real-Time, Thermo Fisher, USA) using TB Green Premix Ex Taq (RR420Q, Takara, Tokyo, Japan). The reaction conditions were as follows: 95C for 10 min, followed by 40 cycles of 95°C for 10 s and 58°C for 60 s. Relative mRNA levels were examined with the 2-ΔΔCt method [22] and normalized against the level of GAPDH. The primers used included: UBC9, forward: 5′-AAAAATCCCGATGGCACGATG-3′, reverse: 5′-CTTCCCACGGAGTCCCTTTC-3′; GAPDH, forward: 5′-GGAGCGAGATCCCTCCAAAAT-3′, reverse: 5′-GGCTGTTGTCATACTTCTCATGG-3′.
Immunoprecipitation (IP) assay
A Pierce Co-Immunoprecipitation kit (26149, Thermo Fisher, USA) was used to examine the interaction between PPARα and SUMO1. Briefly, siUBC9-transfected THP-1 cells were lysed using IP Lysis/Wash Buffer. After being precleared by Agarose Resin, the obtained lysates were centrifuged (2000×g) at 4°C for 20 min to harvest supernatant. Agarose resin-coupled PPARα antibody (ab227074, 1 : 100, Abcam, Cambridge, UK) or normal rabbit IgG (ab171870, Abcam, UK) was used to precipitate target proteins in the supernatant. After overnight incubation at 4°C, the immunocomplexes were eluted using elution buffer (21009, Thermo Fisher, USA) for 5 min and finally detected by Western blot to analyze the number of PPARα and SUMO1 proteins.
Cell treatment
HL-60 and THP-1 cells were treated with 10 µM PPARα inhibitor GW6471 (sc-300779, Santa Cruz Biotechnology, Santa Cruz, CA, USA) pre-dissolved by dimethyl sulfoxide (sc-358801, Santa Cruz Biotechnology, USA) in their culture medium at 37°C for 24 and 48 h [17]. The PPARα agonist pirinixic acid (S8029, 10 µM, Selleckchem, Houston, TX, USA) pre-dissolved in ethanol was applied to incubate transfected/nontransfected THP-1 cells at 37°C for 16 h [17]. Untreated cells acted as the control group.
Viability assay
HL-60 and transfected/nontransfected THP-1 cells (2 × 103) were seeded in each well of 96-well plates and subjected to the treatment mentioned above. Each well was then filled with Cell Counting Kit (CCK)-8 reagent (10 µl, CA1210, Solarbio, Beijing, China) and incubated for 2 h at 37°C. The absorbance at 450 nm was read using a microplate reader (EMax Plus, Molecular Devices, Sunnyvale, CA, USA).
Apoptosis assay
HL-60 and transfected/nontransfected THP-1 cells were treated as mentioned above, and their apoptosis rate was determined employing Annexin V-FITC Early Apoptosis Detection kits (#6592, Cell Signaling Technology, Danvers, MA, USA). Briefly, the treated cells (1 × 106) were harvested via centrifugation at 2000×g for 10 min. After being washed with pre-cooled phosphate buffered saline (PBS; #9808, Cell Signaling Technology, USA), the cells were resuspended in Annexin-V binding buffer. The cell suspension (96 µl) was incubated with 1 µl of Annexin V-FITC and 12.5 µl of propidium iodide solution for 10 min in the dark. Finally, the stained cells were again suspended with Annexin-V binding buffer and analyzed using a flow cytometer (Cytoflex, Beckman Coulter, Brea, CA, USA).
BODIPY-C11 staining
Following the above-mentioned treatment, HL-60 and transfected/nontransfected THP-1 cells were stained with 5 µM BODIPY-C11 (D3861, Thermo Fisher, USA) in 1 ml of their media for 20 min at 37°C. Excess stain was then removed by washing with PBS, and the cells were filtered using a 0.4 µm nylon filter. The activity of lipid peroxidation was reflected by the level of lipid reactive oxygen species (ROS), and its fluorescence was detected by flow cytometry.
Fe2+ level assessment
Intracellular levels of Fe2+ were measured using iron kits (ab83366, Abcam, USA). HL-60 and transfected/nontransfected THP-1 cells (2 × 105/well) were plated in 6-well plates, treated as described above, and reacted with assay buffer for 30 min at 37°C. Then, after incubation with 1 µmol Fe2+ probes for 60 min at 37°C, the cells were subjected to absorbance detection by the microplate reader at 593 nm.
Western blot
HL-60 and transfected/nontransfected THP-1 cells following the above-mentioned treatment were lysed using RIPA Lysis Buffer (20-188, Sigma-Aldrich, St. Louis, MO, USA) to isolate total protein. Then, the protein concentration was determined using the bicinchoninic acid kit (A53227, Thermo Fisher, USA). After being denatured by boiling at 95°C for 5 min, 30 µg of isolated proteins were separated on sodium dodecyl sulfate polyacrylamide gel electrophoresis gel (1615100, BIO-RAD, Hercules, CA, USA), electrophoretically transferred onto a polyvinylidene fluoride (PVDF) membrane (1620256, BIO-RAD, USA), and blocked in 5% skim milk for 1 h at room temperature. Primary antibodies for PPARα (ab227074, 52 kDa, 1 : 500, Abcam, UK), SUMO1 (ab32058, 12 kDa, 1 : 1000, Abcam, UK), SLC7A11 (ab175186, 55 kDa, 1 : 1000, Abcam, UK), GPX4 (ab125066, 17 kDa, 1 : 1000, Abcam, UK) and GAPDH (ab8245, 37 kDa, 1 : 500, Abcam, UK) were used to probe the membrane overnight at 4°C. The membrane was then washed three times with TWEEN-20 PBS (28352, Thermo Fisher, USA), followed by incubation with goat anti-rabbit IgG secondary antibodies (ab97051, Abcam, UK) for 2 h at room temperature. On an imaging system (LAS-3000, Fujifilm, Tokyo, Japan), proteins were developed with Clarity Western ECL Substrate (1705060, BIO-RAD, USA). The grayscale value of each protein band was determined using ImageJ software (3.0 version, National Institutes of Health, Bethesda, MA, USA).
Statistical analysis
All statistical values were presented as mean ± standard deviation (SD) from experiments repeated three times. Statistical analysis was performed using GraphPad Prism (version 8.0, GraphPad Software Inc., San Diego, CA, USA). Data were compared among multiple experimental groups via one-way analysis of variance (ANOVA), followed by the Tukey post hoc test. The statistical significance level was set at p < 0.05.
Results
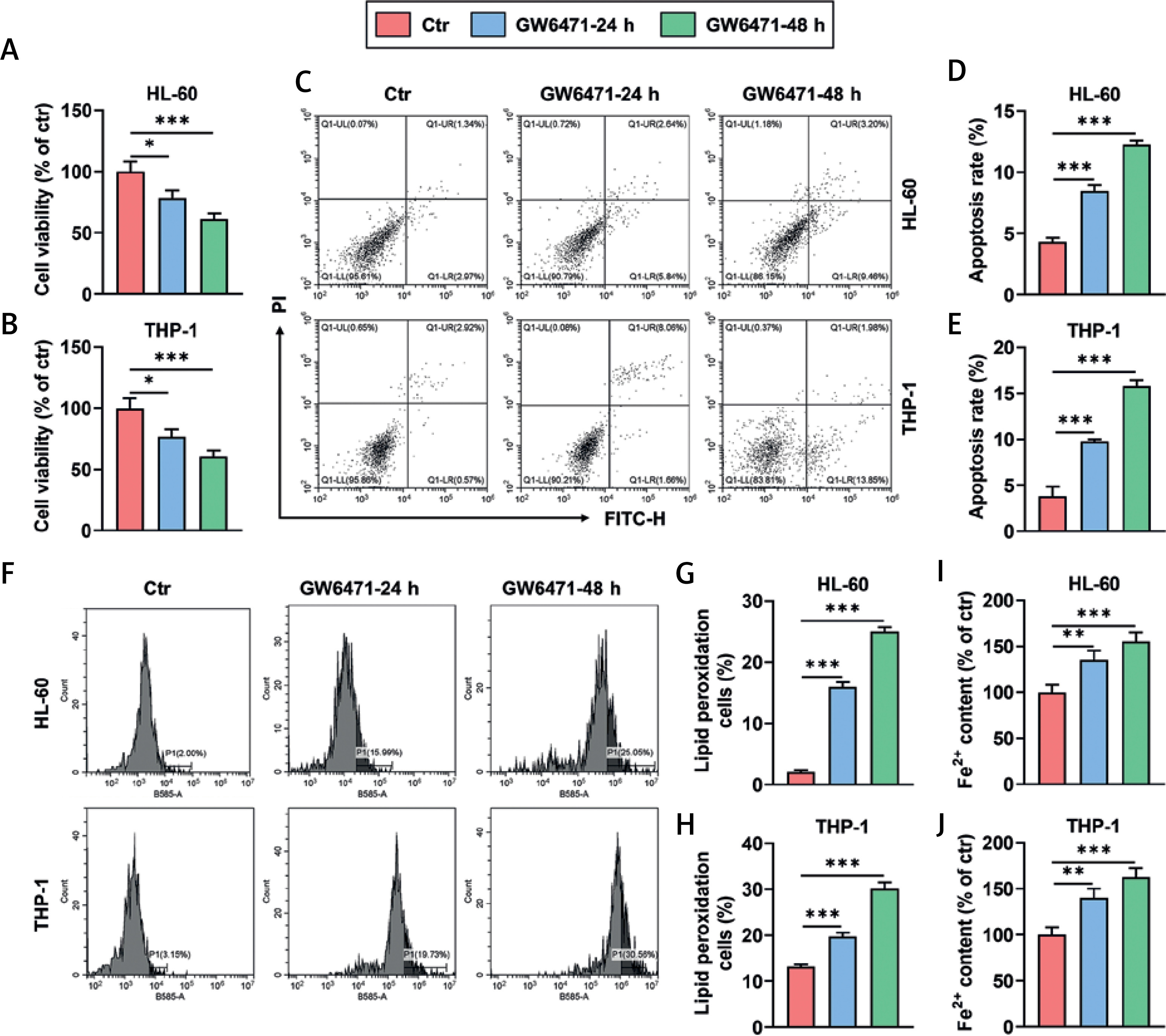
GW6471 treatment for 24 and 48 h decreased viability and promoted apoptosis and ferroptosis of HL-60/THP-1 cells
Treatment with the PPARα inhibitor GW6471 for 24 and 48 h led to decreased viability (Figures 1 A, B, p < 0.05), markedly promoted apoptosis (Figures 1 C–E, p < 0.001), and enhanced activity of lipid peroxidation in HL-60 and THP-1 cells (Figures 1 F–H, p < 0.001). Also, HL-60 and THP-1 cells treated with GW6471 for 24 and 48 h exhibited an increased level of Fe2+ (Figures 1 I, J, p < 0.01).
Figure 1
GW6471 treatment for 24 and 48 h decreased viability and promoted apoptosis and ferroptosis of HL-60/THP-1 cells (A–H). HL-60/THP-1 cells were treated with the PPARα inhibitor GW6471 (10 μM), for 24 and 48 h (A, B). Cell viability was measured by Cell Counting Kit-8 assay (C–E). Cell apoptosis was determined by flow cytometry with Annexin V-FITC/PI staining (F–H). The activity of lipid peroxidation of cells was determined by flow cytometry with BODIPY-C11 staining (I, J). The level of Fe2+ in cells was assessed by colorimetric assay. The values at the ends of the horizontal lines were compared as indicated: *P < 0.05; **P < 0.01; ***P < 0.001 (Ctr, Control)
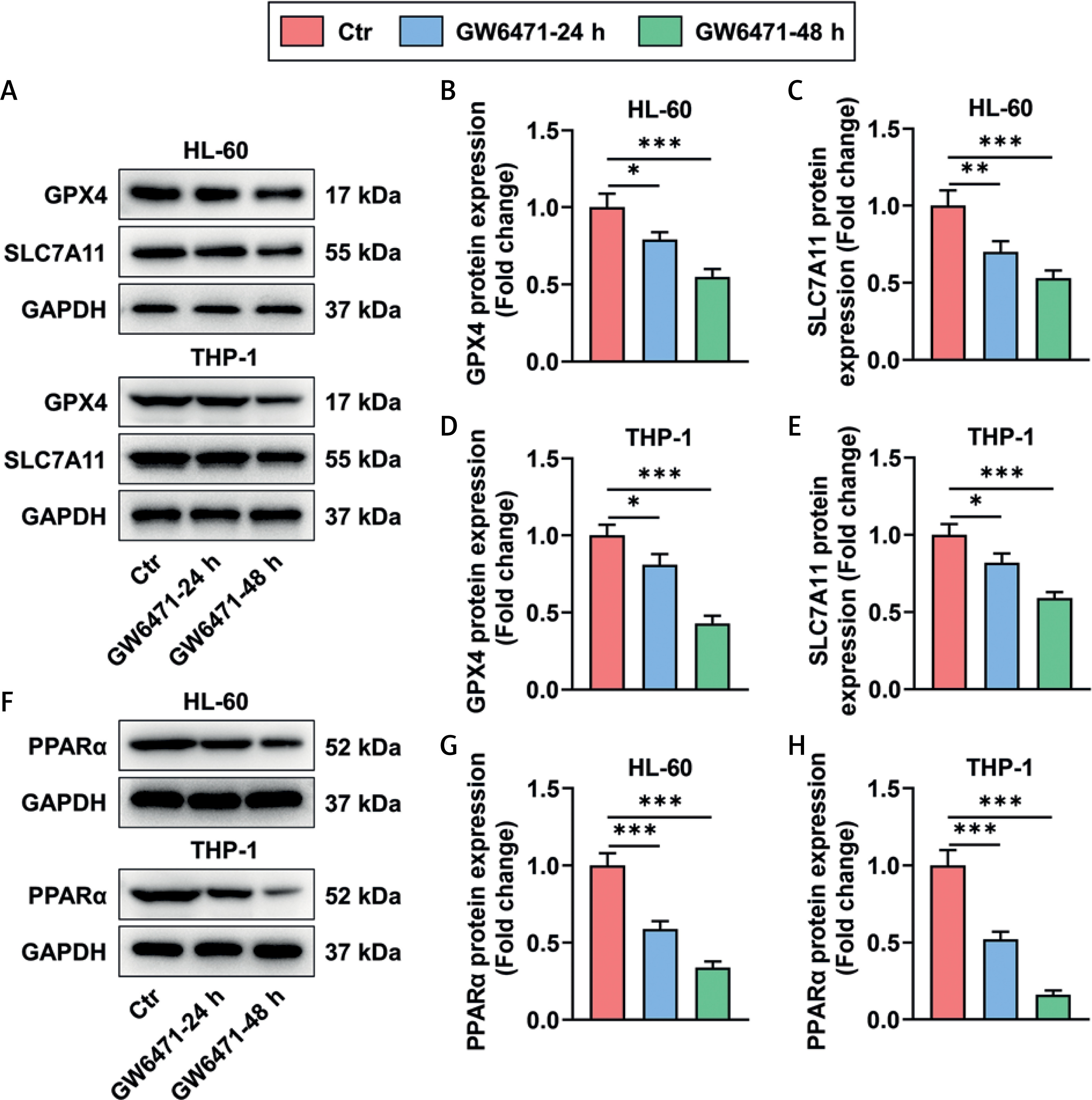
GW6471 treatment for 24 and 48 h reduced the expression of ferroptosis inhibition-related proteins and PPARα in HL-60/THP-1 cells
Expression levels of the ferroptosis inhibition-related proteins GPX4 and SLC7A11 were decreased in HL-60 and THP-1 cells after 24 and 48 h of GW6471 treatment (Figures 2 A–E, p < 0.05). At 24 and 48 h after GW6471 treatment, HL-60 and THP-1 cells presented evident downregulation of PPARα expression (Figures 2 F–H, p < 0.001).
Figure 2
GW6471 treatment for 24 and 48 h reduced the expression of ferroptosis inhibition-related proteins and PPARα in HL-60/THP-1 cells (A–H). The expression levels of GPX4, SLC7A11, and PPARα in HL-60/THP-1 cells treated with the PPARα inhibitor GW6471 (10 μM), for 24 and 48 h, were analyzed by Western blot, with GAPDH used as the normalizer. The values at the ends of the horizontal lines were compared as indicated: *P < 0.05; **P < 0.01; ***P < 0.001 (GPX4, glutathione peroxidase 4; PPARα, peroxisome proliferator-activated receptor α)
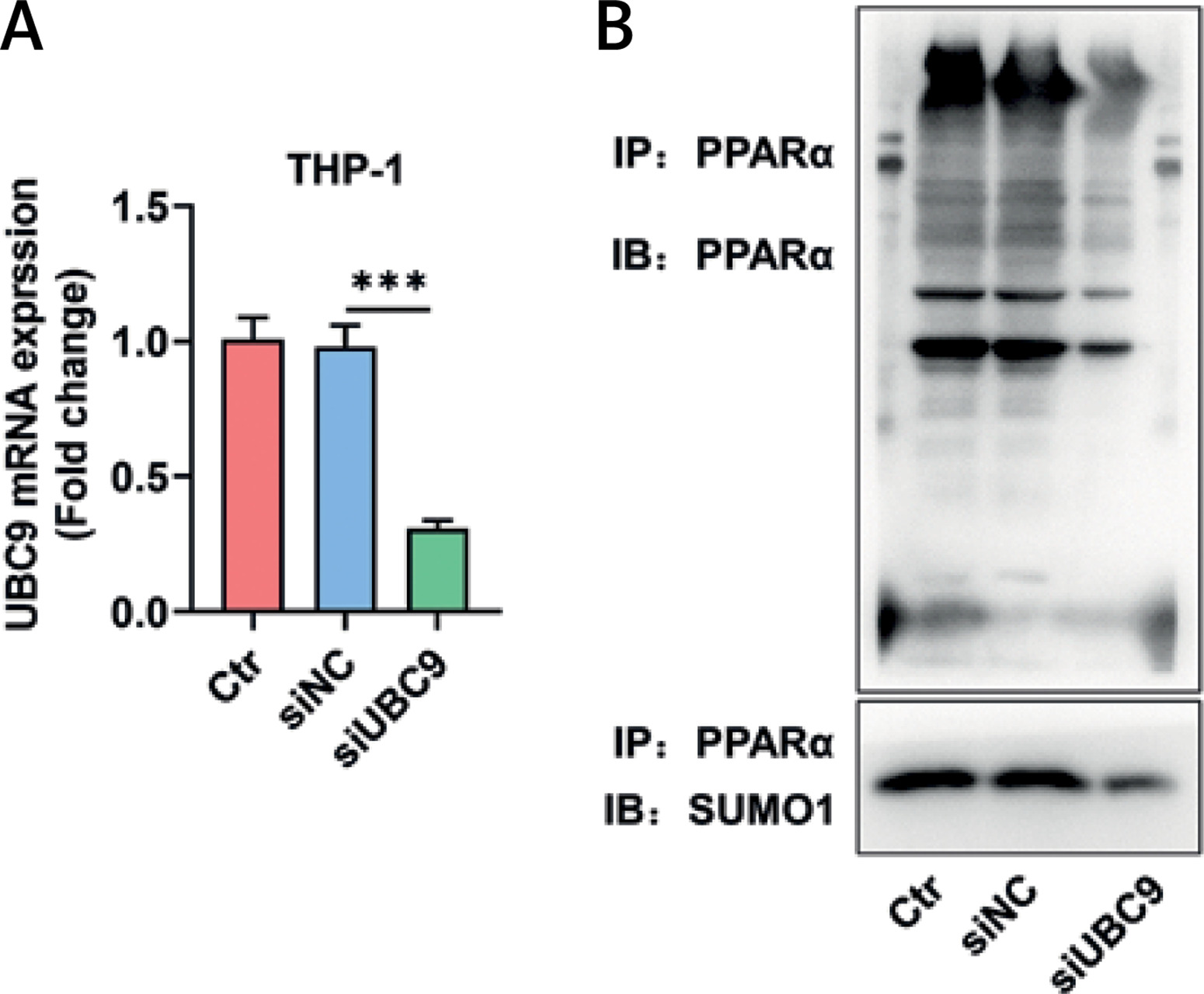
UBC9 silencing attenuated PPARα antibody-induced enrichment of PPARα and SUMO1 in THP-1 cells
Since THP-1 cells showed stronger lipid peroxidation activity than HL-60 cells after GW6471 treatment, THP-1 cells were used in the subsequent experiments. By transfection with siUBC9, UBC9, the only E2 SUMO-conjugating enzyme, was silenced in THP-1 cells (Figure 3 A, p < 0.001). IP assay was conducted to verify the UBC9-mediated SUMOylation of PPARα. As shown in Figure 3 B, PPARα antibodies precipitated both PPARα protein and SUMO1 protein in siNC-transfected THP-1 cells, but precipitated a smaller number of these two proteins in siUBC9-transfected THP-1 cells (Figure 3 B).
Figure 3
UBC9 silencing attenuated the PPARα antibody-induced enrichment of PPARα and SUMO1 in THP-1 cells. A – The expression of UBC9 in si-UBC9/siNC-transfected THP-1 cells was analyzed by qRT-PCR, with GAPDH used as the normalizer. B – The interaction between PPARα and SUMO1 was determined by immunoprecipitation assay in siUBC9/siNC-transfected THP-1 cells. The values at the ends of the horizontal lines were compared as indicated: *** P < 0.001 UBC9 – ubiquitin carrier protein 9, siUBC9 – small interfering RNA targeting UBC9, siNC – small interfering RNA targeting negative control, PPARα – peroxisome proliferator-activated receptor a, SUMO1 – small ubiquitin-related modifier 1, qRT-PCR – quantitative reverse transcription polymerase chain reaction, Ctr – Control.
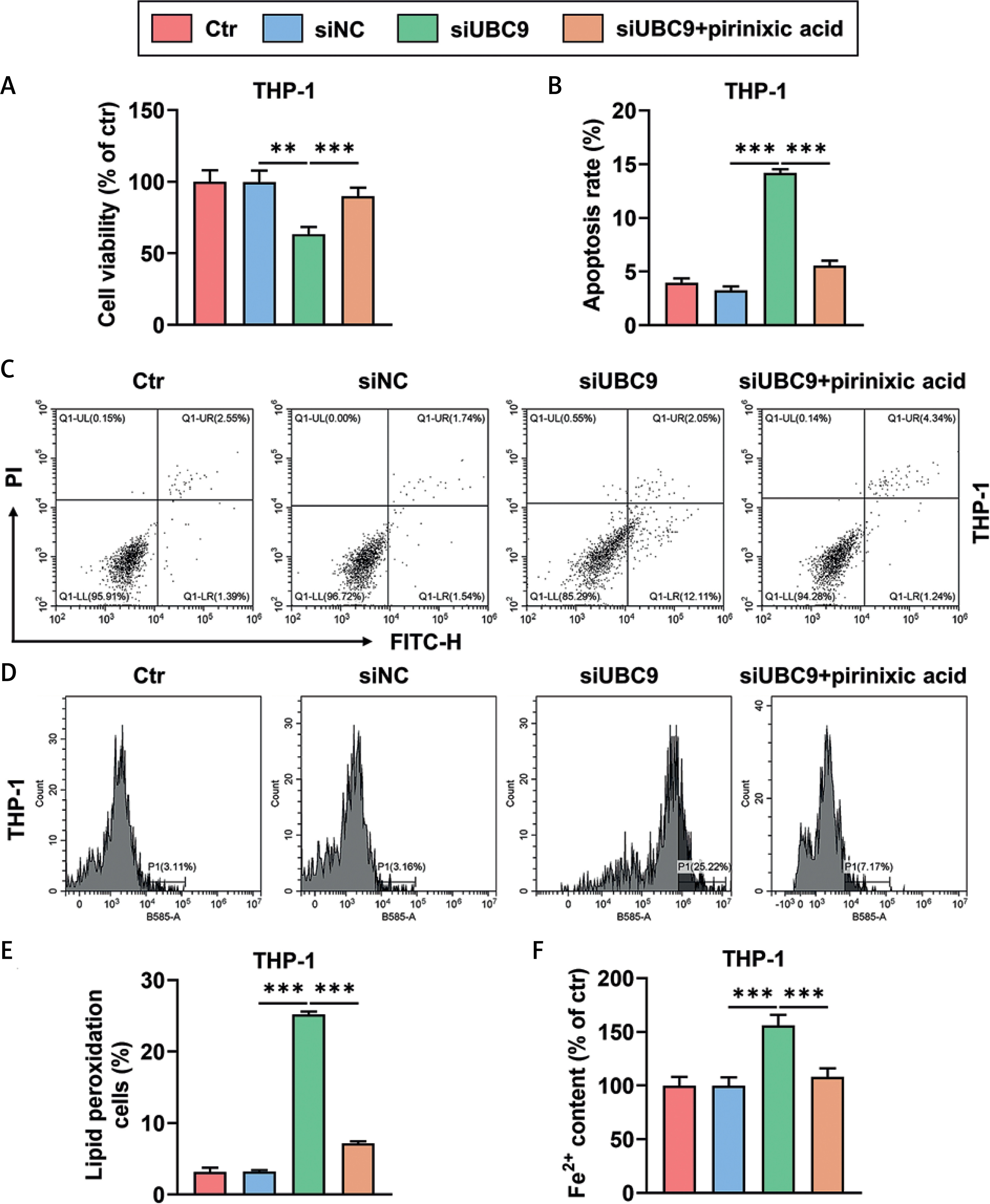
UBC9 silencing-induced viability decrease and promotion of apoptosis and ferroptosis of THP-1 cells were counteracted by pirinixic acid
UBC9 silencing reduced the viability of THP-1 cells (Figure 4 A, p < 0.01), which however was counteracted by the PPARα agonist pirinixic acid (Figure 4 A, p < 0.001). Also, THP-1 cells transfected with siUBC9 showed an increased rate of apoptosis (Figures 4 B, C, p < 0.001), and this siUBC9-induced apoptosis promotion was mitigated by pirinixic acid (Figures 4 B, C, p < 0.001). Moreover, UBC9 silencing augmented the activity of lipid peroxidation and the level of Fe2+ in THP-1 cells (Figures 4 D–F, p < 0.001), which were reversed by pirinixic acid (Figures 4 D–F, p < 0.001).
Figure 4
UBC9 silencing-induced viability decrease and promotion of apoptosis and ferroptosis of THP-1 cells were counteracted by pirinixic acid (A–F). THP-1 cells were transfected with siUBC9/siNC followed by incubation with/ without the PPARα agonist pirinixic acid (10 μM) for 16 h. A – Cell viability was measured by cell counting kit-8 assay. B, C – Cell apoptosis was determined by flow cytometry with Annexin V-FITC/PI staining. D, E – The activity of lipid peroxidation of cells was evaluated by flow cytometry with BODIPY-C11 staining. F – The level of Fe2+ in cells was assessed by colorimetric assay. The values at the ends of the horizontal lines were compared as indicated: **P < 0.01; ***P < 0.001
UBC9 – ubiquitin carrier protein 9, siUBC9 – small interfering RNA targeting UBC9, siNC – small interfering RNA targeting negative control, qRT-PCR – quantitative reverse transcription polymerase chain reaction, Ctr – Control.
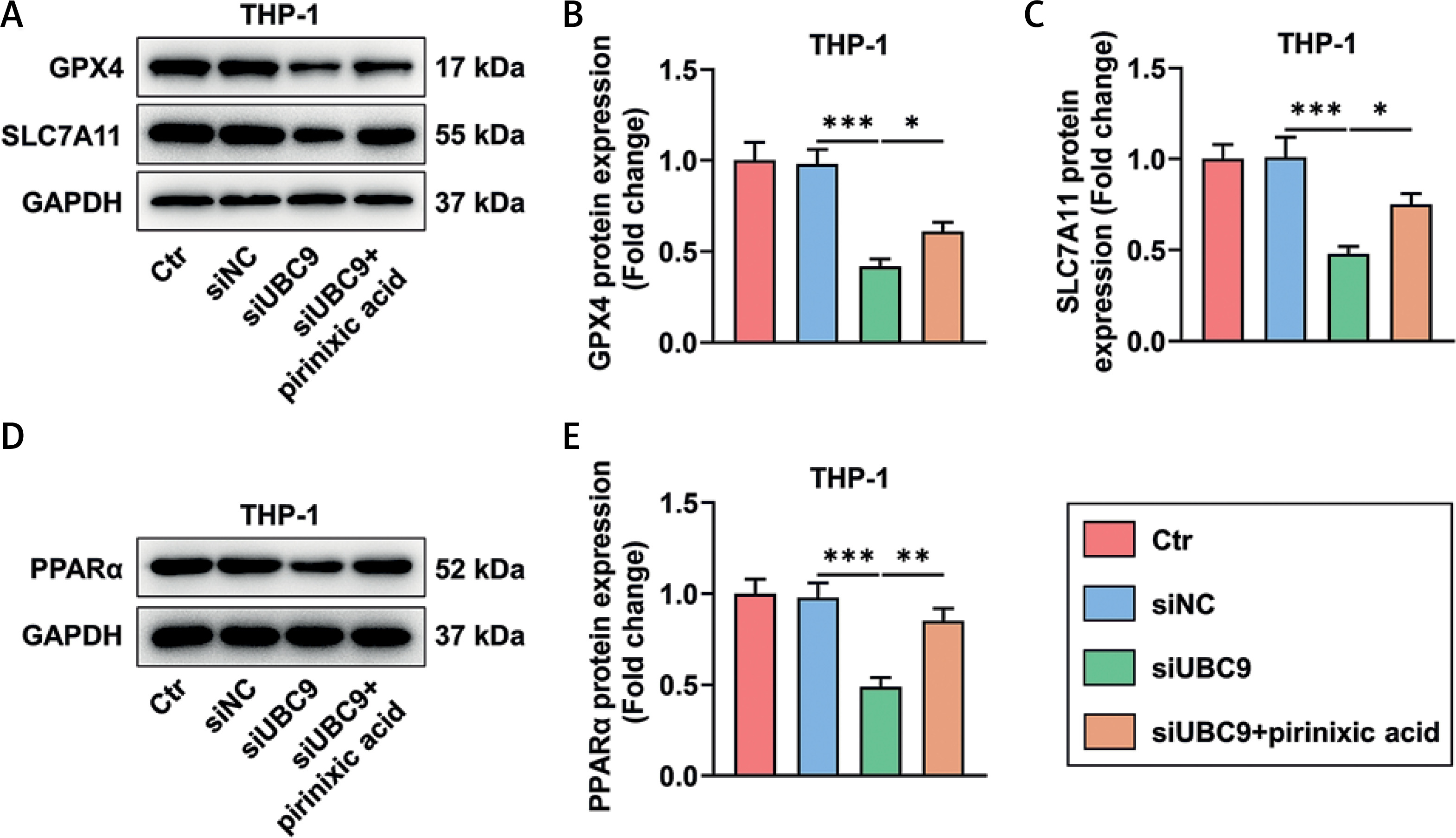
UBC9 silencing-induced GPX4, SLC7A11, and PPARα downregulation was counteracted by pirinixic acid
The expression levels of GPX4 and SLC7A11 as well as PPARα were diminished in THP-1 cells due to UBC9 silencing (Figures 5 A–E, p < 0.001), while such downregulation of GPX4, SLC7A11, and PPARα in THP-1 cells was neutralized partly by pirinixic acid (Figures 5 A–E, p < 0.05).
Figure 5
UBC9 silencing-induced GPX4, SLC7A11, and PPARα downregulation was counteracted by pirinixic acid (A–E). The expression levels of GPX4, SLC7A11, and PPARα in THP-1 cells transfected with siUBC9/siNC followed by incubation with/without the PPARα agonist pirinixic acid (10 μM) for 16 h were analyzed by Western blot, with GAPDH used as the normalizer. The values at the ends of the horizontal lines were compared as indicated: *P < 0.05; **P < 0.01; ***P < 0.001
UBC9 – ubiquitin carrier protein 9, siUBC9 – small interfering RNA targeting UBC9, siNC – small interfering RNA targeting negative control, GPX4 – glutathione peroxidase 4, PPARα – peroxisome proliferator-activated receptor α, Ctr – Control.
Discussion
Despite the recent approval of novel promising therapies for AML, treatment of AML still remains a challenge due to frequent disease relapse [5, 23]. Relapse arises from unhindered cell proliferation, which is often associated with cytogenetic abnormalities in AML [24]. Targeting these abnormalities can contain the progression of AML [25]. Moreover, genes involved in ferroptosis signaling pathways have been reported to impact AML patients’ prognosis [26]. The present study revealed that destabilizing PPARα can promote ferroptosis to inhibit AML cell proliferation.
PPARα, the first identified member of the NR1C nuclear receptor subfamily, is predominantly expressed in the liver [27]. PPARα can be activated by peroxisome proliferators, which elicit peroxisome proliferation and liver cancer in mice [28]. PPARα contributes to mono(2-ethylhexyl) phthalate-caused acceleration of ovarian cancer development [29]. Conversely, intestinal PPARα has been recorded to protect mice against colon carcinogenesis [30]. Previously, Luo et al. reported that the ubiquitin-dependent proteasomal degradation of PPARα by Tribbles homolog 3 reduces apoptosis and autophagy of AML cells, implying that PPARα plays a tumor-suppressive role in AML [31]. Notably, contrary to Luo’s finding, our study showed that PPARα inhibition caused by GW6471 treatment reduced AML/THP-1 cell viability and enhanced cell apoptosis, suggesting that PPARα was oncogenic in AML. Our results are supported by Li’s study where PPARα expression was downregulated when ATGL silencing induced inhibition of AML cell proliferation [14].
In response to ligand stimulation, PPARα functions as a transcriptional program that greatly augments fatty acid uptake and breakdown through fat acid oxidation [32]. PPARα can act as a transcriptional activator inducing multidrug resistance-associated protein 2 (Mdr2) expression to mediate hepatobiliary cholesterol transport in mice [33]. By enhancing PPARα activity, pharmacological fibrates are able to lower triglyceride [34]. In addition, PPARα activation by fasting promotes hepatic fatty acid oxidation and gluconeogenesis [28]. Ferroptosis is a form of programmed cell death, the sensitivity of which can be increased with accumulation of oxidized cellular membrane phospholipids [35]. Since PPARα enhances fatty acid oxidation to reduce lipid accumulation [36], PPARα is expected to antagonize ferroptosis, which is corroborated by the previous finding that PPARα activation mitigates erastin-induced liver cancer cell ferroptosis [17]. Ferroptosis occurs when the Fenton reaction induces iron-dependent lethal lipid peroxidation, and Fe2+ is needed for conversion into Fe3+ to run the reaction [37]. GPX4 is an enzyme preventing the toxicity of lipid peroxides to maintain the homeostasis of membrane lipid bilayers [38]. To initiate ferroptosis, GPX4 inactivation is caused by RSL3, leading to the accumulation of lipid peroxides [6]. SLC7A11 is a key component of system xc−, through which extracellular cystine is transported into cells by exchange of intracellular glutamate and then converted into cysteine, and cysteine is required for the generation of glutathione, whose depletion also gives rise to GPX4 inactivation [39]. Also, repression of SLC7A11 impairs cysteine import to deplete glutathione, thereby inactivating GPX4 to trigger ferroptosis [40]. Ferroptosis is more immunogenic than apoptosis in causing cell death [7], and stimulating the ferroptotic pathway may contribute to immunotherapy against cancers including AML [41, 42]. Promoting ferroptosis by all-trans retinoic acid derivative has been proven to facilitate AML cell differentiation and thereby prevent AML progression [43]. Our study provided a new finding that GW6471-caused PPARα inhibition boosted ferroptosis of AML/THP-1 cells by elevating lipid peroxidation and Fe2+ levels and inhibiting the production of GPX4 and SLC7A11, suggesting that repressing PPARα activity can retard AML cell growth.
Sumoylation, a type of reversible post-translational modification, can modify genes to alter their functions in AML, thus affecting AML progression [21]. DeSUMOylation leads to ubiquitination-mediated degradation of PPARα [20]. UBC9 is a SUMO-conjugating enzyme that transfers activated small SUMOs such as SUMO1 to various protein substrates [44], preceding attachment of SUMO to the protein by an E3 ligase [19]. Our IP assay confirmed that UBC9 silencing destabilized PPARα by preventing SUMO1 from ligating PPARα to deSUMOylating PPARα in THP-1 cells. Knockdown of UBC9 has been reported to inhibit AML cell proliferation [21], which is similar to our finding that UBC9 silencing, like PPARα inhibition, resulted in decreased cell viability and levels of GPX4, SLC7A11, and PPARα, and promotion of apoptosis and ferroptosis in THP-1 cells. Furthermore, increasing PPARα activity was found to reverse all the above effects of UBC9 silencing. Our results taken together indicated that UBC9 silencing deSUMOylated PPARα to induce PPARα inhibition, which inhibited AML cell growth by facilitating ferroptosis. Our study highlighted the potential of the ferroptosis pathway as a therapeutic target for AML, not only improving the understanding of ferroptosis, but also providing potential new therapeutic strategies for the clinical treatment of AML. Nonetheless, there were some limitations and shortcomings. Given only investigation at the cellular level, further validation of the results in animal studies is necessary.
In conclusion, the present study demonstrated that UBC9 silencing-induced deSUMOylation results in PPARα inhibition, whereby ferroptosis is facilitated to suppress AML cell growth. Our findings provide a rationale for AML treatment with a PPARα inhibitor.